La resolución del caso!
Varón de 38 años con dolor abdominal, ictericia y disnea. ¡Lo invitamos a opinar!
Datos personales: varón de 38 años
Motivo de consulta: dolor abdominal, ictericia y disnea
Enfermedad actual: Comienza 5 días previos a la consulta con coloración amarillenta de la piel, generalizada, dolor abdominal continuo de intensidad moderada, localizado predominantemente en hipocondrio derecho asociado a distensión abdominal.
Del mismo tiempo de evolución refiere debilidad generalizada, astenia, disfonía, odinofagia y tos seca persistente.
Enfermedad actual: Comienza 5 días previos a la consulta con coloración amarillenta de la piel, generalizada, dolor abdominal continuo de intensidad moderada, localizado predominantemente en hipocondrio derecho asociado a distensión abdominal.
Del mismo tiempo de evolución refiere debilidad generalizada, astenia, disfonía, odinofagia y tos seca persistente.
El día previo agrega disnea ante moderados esfuerzos la cual progresa hasta hacerse de reposo.
Antecedentes Personales:
- Hace 2 años comienza estudio de forma ambulatoria por el servicio de Dermatología por dermatosis en ambos miembros inferiores de un año de evolución descriptas en la historia clínica como lesiones costrosas con gran eccema y algunas vesículas en la periferia que evoluciona con exacerbaciones y remisiones espontáneas.
- Se realiza una biopsia de piel de dichas lesiones que informa: Infiltrado inflamatorio constituido por linfocitos y eosinófilos en menor proporción perivascular de vasos superficiales y profundos con compromiso de anexos cutáneos. El proceso se extiende en algunas áreas al intersticio donde además se hallan mastocitos. Las características morfológicas corresponden a un patrón de reacción tipo granuloma de Churg Strauss.
- Comienza tratamiento con Prednisona 40 mg/día con leve mejoría de las lesiones pero sin remisión completa.
- Herpes Zóster torácico hace 2 años para el cual realizó tratamiento con aciclovir.
- Alcohol: 40 g/día, durante 18 años
- Tabaco: 5 paquetes/año
Examen físico:
Vigil, globalmente orientado.
Vigil, globalmente orientado.
Signos vitales:
- Presión arterial: 120/70 mmHg
- Frecuencia cardíaca: 120 latidos/minuto
- Frecuencia respiratoria: 36 ciclos/minuto
- Temperatura: 37° C
Cabeza y cuello: conjuntivas congestivas, escleras ictéricas; piezas dentarias incompletas y otras en mal estado. Orofaringe muy congestiva, con puntillado petequial en paladar. Se palpan adenopatías duro elásticas dolorosas móviles (1 x 1 cm) submaxilares, submandibulares y axilares bilaterales.
Tórax: lesiones maculares eritematosas de bordes netos en dorso de 6 x 5 cm.
Tórax: lesiones maculares eritematosas de bordes netos en dorso de 6 x 5 cm.
Aparato respiratorio: Respiración costoabdominal superficial. Matidez en base izquierda. Murmullo vesicular disminuido en base pulmonar izquierda, sin ruidos agregados.
Aparato cardiovascular: Ruidos hiperfonéticos; sin R3 ni R4, ni soplo.
Abdomen: distendido, sin cicatrices ni circulación colateral. Ruidos hidroaéreos presentes. Tenso, doloroso a la palpación profunda en hemiabdomen derecho. Matidez desplazable. Hepatomegalia a 7 cm del reborde costal derecho, esplenomegalia leve. Ruidos hidroaéreos presentes.
Miembros: trofismo disminuido, temperatura aumentada. Lesiones maculares eritematosas con edema duro en ambos miembros inferiores. Ampollas en dorso de pie izquierdo. Se palpa adenopatía inguinal izquierda 2 x 3 cm y derecha 1 x 2 cm
Exámenes complementarios:
Abdomen: distendido, sin cicatrices ni circulación colateral. Ruidos hidroaéreos presentes. Tenso, doloroso a la palpación profunda en hemiabdomen derecho. Matidez desplazable. Hepatomegalia a 7 cm del reborde costal derecho, esplenomegalia leve. Ruidos hidroaéreos presentes.
Miembros: trofismo disminuido, temperatura aumentada. Lesiones maculares eritematosas con edema duro en ambos miembros inferiores. Ampollas en dorso de pie izquierdo. Se palpa adenopatía inguinal izquierda 2 x 3 cm y derecha 1 x 2 cm
Exámenes complementarios:
Laboratorio general
.
|
Ingreso
|
Día 2º
|
Día 3º
|
Día 4º
|
Día 5º
|
| Hematocrito (%) | 40 | 33 | 36 | 32 | 22 |
| Hemoglobina (g/dl) | 13,5 | 11 | 12 | 11 | 7 |
| Leucocitos/mm3 | 1.100 | 1.170 | 1.400 | 1.700 | 2.800 |
| Plaquetas/mm3 | 21.000 | 7.000 | 18.000 | 14.000 | 97.000 |
| Glicemia (mg/dl) | 89 | 120 | 53 | 54 | 56 |
| Tiempo de protrombina (segundos) Tasa de Protrombina (%) | 28,3 25 | . | 34 20 | 23 34 | |
| KPPT (segundos) | 59 | . | 54 | 43 | |
| Urea (mg/dl) | 95 | 97 | 110 | 160 | 230 |
| Creatininemia (mg/dl) | 1,5 | 1,5 | 1,7 | 3,2 | 4,3 |
| Proteínas totales (g/dl) | 4,8 | 4,1 | |||
| Albúmina (g/dl) | 2,3 | 2,2 | |||
| TGO (UI/l) | 351 | 358 | 360 | 430 | 530 |
| TGP (UI/l) | 203 | 494 | 440 | 341 | 342 |
| Fosfatasa alcalina (UI/l) | . | 1615 | 1797 | 1410 | 1404 |
| GGT (UI/l) | . | 292 | 300 | 305 | |
| Colinesterasa sérica | 3605 | . | 2783 | ||
| Bilirrubina total (mg/dl) | 19 | 20 | 22 | 20 | |
| Bilirrubina directa (mg/dl) | 15 | 16 | 17 | 18 | |
| Bilirrubina indirecta (mg/dl) | 4 | 4 | 5 | 2 | |
| Natremia (mEq/l) | 129 | 125 | 126 | 128 | 125 |
| Potasemia (mEq/l) | 4,1 | 5,1 | 4,9 | 5,1 | 6 |
| Láctico deshidrogenasa (UI/l) | . | 3078 | . | ||
| Velocidad de eritrosedimentación (mm/1º hora) | 2 | . | . | ||
| EAB (FIO2 %) | 21 | . | . | 28 | 50 |
| PH | 7,35 | . | . | 7,27 | 6,8 |
| PaO2 (mmHg) | 24 | . | . | 27 | 24 |
| PaCO2 (mmHg) | 63 | . | . | 67 | 93 |
| EB (mmol/L) | -10 | . | . | -12 | -29 |
| Bicarbonato estándar (mmol/L) | 16 | . | . | 14 | 5 |
| Bicarbonato real (mmol/L) | 13 | . | . | 12 | 4 |
| % Saturación | 89 | . | . | 89 | 86 |
Orina completa (Ingreso):
Aspecto: turbio, color: naranja, pH: 5, densidad: 1030, proteínas: 1,58 g/I, glucosa: no contiene, cuerpos cetónicos: no contiene, pigmentos biliares: +++, urobilinas: normal, hemoglobina: ++
Sedimento urinario:
- Hematíes: abundantes, eumórficos
- Leucocitos: escasos
- Células epiteliales: escasas
- Piocitos: no contiene
- Cilindros: 1/ 1-2 campos, hialinos
Radiografía tórax frente (Ingreso): derrame pleural izquierdo leve. Tenue infiltrado intersticial bilateral
Electrocardiograma: taquicardia sinusal; resto sin alteraciones
Análisis cito-físico-químico de líquido pleural (ingreso) Glucosa: 1,12g/I, proteínas: 20 g/I, albúmina: 7 g/I, colesterol total: 15 mg/dl, triglicéridos: 27 mg/dl, colinesterasa: 15 UI/I, amilasa: 34UI/I, LDH: 1354 UI/I, pH: 7,41, reacción de Rivalta: negativo, recuento de elementos: 800 /mm3, predominio de PMN.
Análisis cito-físico-químico de líquido ascítico (ingreso) Glucosa: 0,93 g/I, proteínas: 19 g/I, albúmina: 0 g/I, colesterol total: 25 mg/dL, triglicéridos: 72 mg/dl, colinesterasa: 1115 UI/I, amilasa: 16 UI/I, LDH: 1151 UI/I, bilirrubina total: 6,68 mg/dl, bilirrubina directa: 5,56 mg/dl, bilirrubina indirecta: 1,06 mg/dl, reacción de Rivalta: ++++, recuento de elementos: 98 /mm3, gradiente seroascítico de albúmina (GASA): 2,2
Análisis cito-físico-químico de líquido ascítico (ingreso) Glucosa: 0,93 g/I, proteínas: 19 g/I, albúmina: 0 g/I, colesterol total: 25 mg/dL, triglicéridos: 72 mg/dl, colinesterasa: 1115 UI/I, amilasa: 16 UI/I, LDH: 1151 UI/I, bilirrubina total: 6,68 mg/dl, bilirrubina directa: 5,56 mg/dl, bilirrubina indirecta: 1,06 mg/dl, reacción de Rivalta: ++++, recuento de elementos: 98 /mm3, gradiente seroascítico de albúmina (GASA): 2,2
Extendido de sangre periférica (Día 2)
- Microhematocrito (%): 39
Morfología de serie roja: normocromía, normocitosis
- Leucocitos/mm3: 1.200 (Neutrófilos en cayado 10%; Neutrófilos segmentados 44%; Linfocitos 22%; Monocitos 24%)
- Plaquetas/mm3: 140.000 (macroplaquetas)
Punción Biopsia de Médula Ósea (Día 2)
Hiperplasia de la serie eritroide, con 11% de células atípicas (por medulograma)
Megacariocitos presentes formadores de plaquetas
Laboratorio inmunológico:- FAN negativo
- Fracciones del complemento: normal
- p-ANCA: negativo
- Factor reumatoideo: reactivo 1/40
- VDRL: no reactivo
Estudios serológicos: → Test de mononucleosis infecciosa (Monotest): no reactivo
→ Ac totales anti T. gondii: reactiva 1/8
→ VIH, HVC, HVB: negativo
Estudios microbiológicos:
→ Urocultivo: negativo
→ Hemocultivos (dos de dos): Pseudomona aeruginosa sensible a ceftazidima, ampicilina-sulbactam, ciprofloxacina, amikacina, piperacilina, imipenem
→ Cultivo de líquido pleural y ascítico: negativo
Ecografía abdominal (Ingreso):
Hígado: aumentado de tamaño. Parénquima difusamente heterogéneo, desatacándose en segmento VI, imagen nodular hipoecogénica de 18 mm aproximadamente y en segmento III imagen de similares características de 14 mm.
Vesícula: colapsada.
Vía biliar y páncreas: sin particularidades.
Bazo: aumentado de tamaño.
Se observa líquido libre en cavidad peritoneal de moderada magnitud.
Tomografía computada de tórax, abdomen y pelvis sin contraste EV (Día 3):- Microhematocrito (%): 39
Morfología de serie roja: normocromía, normocitosis
- Leucocitos/mm3: 1.200 (Neutrófilos en cayado 10%; Neutrófilos segmentados 44%; Linfocitos 22%; Monocitos 24%)
- Plaquetas/mm3: 140.000 (macroplaquetas)
Punción Biopsia de Médula Ósea (Día 2)
Hiperplasia de la serie eritroide, con 11% de células atípicas (por medulograma)
Megacariocitos presentes formadores de plaquetas
Laboratorio inmunológico:- FAN negativo
- Fracciones del complemento: normal
- p-ANCA: negativo
- Factor reumatoideo: reactivo 1/40
- VDRL: no reactivo
Estudios serológicos: → Test de mononucleosis infecciosa (Monotest): no reactivo
→ Ac totales anti T. gondii: reactiva 1/8
→ VIH, HVC, HVB: negativo
Estudios microbiológicos:
→ Urocultivo: negativo
→ Hemocultivos (dos de dos): Pseudomona aeruginosa sensible a ceftazidima, ampicilina-sulbactam, ciprofloxacina, amikacina, piperacilina, imipenem
→ Cultivo de líquido pleural y ascítico: negativo
Ecografía abdominal (Ingreso):
Hígado: aumentado de tamaño. Parénquima difusamente heterogéneo, desatacándose en segmento VI, imagen nodular hipoecogénica de 18 mm aproximadamente y en segmento III imagen de similares características de 14 mm.
Vesícula: colapsada.
Vía biliar y páncreas: sin particularidades.
Bazo: aumentado de tamaño.
Se observa líquido libre en cavidad peritoneal de moderada magnitud.
1. Infiltración parenquimatosa pulmonar bilateral a predominio central y lóbulos superiores, con áreas de opacificación en vidrio esmerilado y engrosamiento de los septos inter e intralobulillares a predominio central asociado a derrame pleural bilateral. Se evidencia alteración de la grasa mediastinal.
2. Hepatoesplenomegalia.
3. Líquido intraabdominal escaso.
4. Impresiona adenomegalias intercavoaórticas (no se puede confirmar por falta de contraste EV).
5. Aumento del tamaño de ambos riñones.
2. Hepatoesplenomegalia.
3. Líquido intraabdominal escaso.
4. Impresiona adenomegalias intercavoaórticas (no se puede confirmar por falta de contraste EV).
5. Aumento del tamaño de ambos riñones.
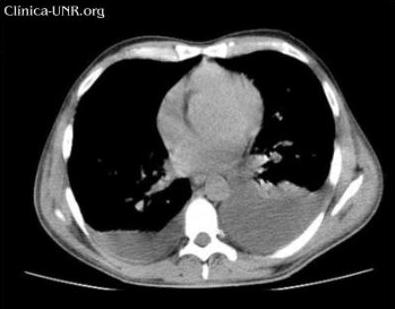
TC de tórax sin contraste EV: derrame pleural bilateral a predomino izquierdo y alteración de la grasa mediastinal
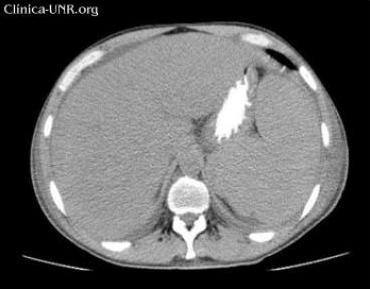
TC de abdomen sin contraste EV: hepatoesplenomegalia marcada. Líquido intraabdominal escaso
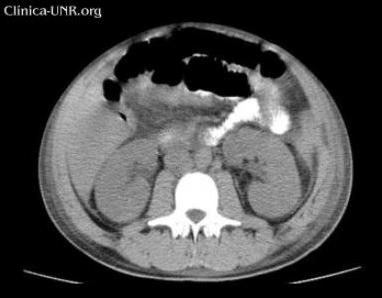
TC de abdomen sin contraste: líquido intraabdominal escaso. Aumento del tamaño de ambos riñones.
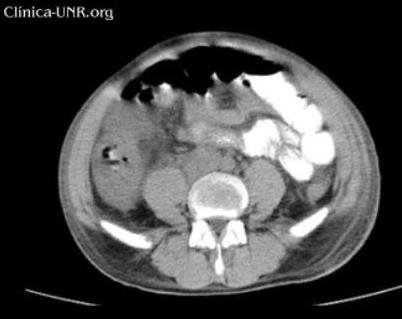
TC de abdomen sin contraste EV: adenomegalias intercavoaórticas, que no se pueden confirmar por la falta de contraste endovenoso. Escaso líquido intraabdominal.
Evolución:
Al ingreso se toman muestras de sangre, orina, líquido pleural y ascítico para cultivos microbiológicos e inicia tratamiento empírico con Piperacilina-Tazobactam y Vancomicina.
Durante la internación persiste con distensión y dolor abdominal en hipocondrio derecho, sin defensa ni descompresión con ruidos hidroaéreos presentes. Se realizan transfusiones de concentrados de plaquetas en días sucesivos.
El día 2º de internación presenta un episodio de enterorragia, gingivorragia y epistaxis por lo que se transfunde nuevamente plaquetas y 6 unidades de plasma fresco.
El día 3º presenta oliguria, empeoramiento de los parámetros de función renal asociado a signos de insuficiencia respiratoria por lo que se decide su traslado a Unidad de Terapia Intensiva. Realiza tratamiento diurético sin respuesta.
Debido a la progresión de la insuficiencia respiratoria el 4º día se realiza intubación orotraqueal y conexión a asistencia mecánica respiratoria. Además inicia hemodiálisis de urgencia.
El día 5º presenta bradicardia extrema y paro cardiorrespiratorio sin respuesta a maniobras de reanimación avanzada.
Discusión del caso clínicoDra. María Lourdes Garibotti (Clínica Médica)
Se discute el caso de un paciente de sexo masculino de 38 años de edad con antecedentes de vasculitis en miembros inferiores diagnosticada hace un año y en tratamiento con prednisona 35 mg/día que consultó por ictericia, dolor abdominal, y disnea progresiva. En el examen físico se constata poliadenopatías, hepatoesplenomegalia y lesiones eritematosas en piel a nivel la pared posterior del tórax. En el laboratorio presenta bicitopenia (leucopenia y plaquetopenia), prolongación del tiempo de protrombina, aumento de transaminasas y de bilirrubina, insuficiencia renal e hipoxemia.
Las preguntas que me planteo ante este caso son las siguientes:
Se discute el caso de un paciente de sexo masculino de 38 años de edad con antecedentes de vasculitis en miembros inferiores diagnosticada hace un año y en tratamiento con prednisona 35 mg/día que consultó por ictericia, dolor abdominal, y disnea progresiva. En el examen físico se constata poliadenopatías, hepatoesplenomegalia y lesiones eritematosas en piel a nivel la pared posterior del tórax. En el laboratorio presenta bicitopenia (leucopenia y plaquetopenia), prolongación del tiempo de protrombina, aumento de transaminasas y de bilirrubina, insuficiencia renal e hipoxemia.
Las preguntas que me planteo ante este caso son las siguientes:
1º) ¿Cuál fue la enfermedad de base de nuestro paciente?
2º) ¿Fueron las lesiones de miembros inferiores un epifenómeno de la enfermedad actual?
3º) ¿Cuál fue la causa de muerte?
Si analizaron las posibles causas que podrían haber provocado la enfermedad de base del paciente, las mismas podrían ser infecciosas, neoplásicas, inmunológicas y otras.
Dentro de las primeras se destacan aquellas producidas por bacterias, hongos y virus. Entre las causas bacterianas que pueden producir este cuadro encontramos al Micobacterium tuberculosis, a las micobacterias atípicas y a la infección por Treponema pallidum. Con respecto a este último, se obtuvo una VDRL negativo por lo cual se descarta el diagnóstico.
La tuberculosis es una enfermedad infectocontagiosa producida por el Micobacterium tuberculosis, que presenta una alta incidencia en nuestro país. Se caracteriza por presentar clínicamente un compromiso pulmonar y extrapulmonar. Este último se puede presentar como tuberculosis ganglionar, pleural, genitourinaria, gastrointestinal y miliar o diseminada. La forma clínica miliar afecta más frecuentemente pulmones, hígado y bazo. Puede afectar además médula ósea, riñón, sistema nervioso, glándulas adrenales y peritoneo.
Los datos a favor para pensar en este diagnóstico son el área endémica, que el paciente recibió durante un año tratamiento inmunosupresor y el cuadro clínico compatible.
En contra del mismo el paciente no presentaba un foco epidemiológico y en el medulograma se observaron células atípicas en un 10 %.
Entre las causas micóticas se destaca la histoplasmosis, coccidiomicosis y paracoccidiomicosis. La primera es una micosis producida por el Histoplasma capsulatum y el contagio es por vía inhalatoria. Puede presentarse de forma aguda constituyendo la forma primaria aguda con síntomas inespecíficos que va desde una enfermedad gripal leve hasta un cuadro compatible con una neumonía clásica o bien de forma progresiva, forma diseminada progresiva, que se caracteriza por presentar compromiso pulmonar, linfadenopatía con hepatoesplenomegalia, lesiones cutáneo mucosas polimorfas, úlceras oro-faríngeas profundas e indoloras. Puede afectar la médula ósea y presentar pancitopenia. Con respecto a estas micosis destaco que los estados de inmunodepresión son un factor predisponente para el contagio de las mismas.
Entre las causas micóticas se destaca la histoplasmosis, coccidiomicosis y paracoccidiomicosis. La primera es una micosis producida por el Histoplasma capsulatum y el contagio es por vía inhalatoria. Puede presentarse de forma aguda constituyendo la forma primaria aguda con síntomas inespecíficos que va desde una enfermedad gripal leve hasta un cuadro compatible con una neumonía clásica o bien de forma progresiva, forma diseminada progresiva, que se caracteriza por presentar compromiso pulmonar, linfadenopatía con hepatoesplenomegalia, lesiones cutáneo mucosas polimorfas, úlceras oro-faríngeas profundas e indoloras. Puede afectar la médula ósea y presentar pancitopenia. Con respecto a estas micosis destaco que los estados de inmunodepresión son un factor predisponente para el contagio de las mismas.
Las causas virales: (VIH, Virus Epstein-Barr, Citomegalovirus) quedan descartadas debido a que las serologías fueron negativas.
Las neoplasias son otros de los diagnósticos que podrían producir la enfermedad de base de este paciente. Las voy a dividir en hematológicas (linfomas, histiocitosis maligna, leucemias agudas) y tumores sólidos.
En cuanto a los linfomas, la enfermedad de Hodgkin es un trastorno linfoproliferativo que presenta un pico epidemiológico bimodal, a los 20 a 30 años y en la quinta década, predomina en varones y presenta cuatro variantes anatomopatológicas: predominio linfocitario, esclerosis nodular, celularidad mixta y depleción linfocitaria. Clínicamente puede manifestarse por adenopatías superficiales o profundas. El bazo sería el lugar de inicio de la enfermedad abdominal y la afectación hepática, de médula ósea y hueso implican diseminación sanguínea. Las infecciones virales, micóticas y oportunistas se asocian a esta patología debido a que hay una alteración de la inmunidad humoral.
El Linfoma no Hodgkin se origina en un 85 a 90% en células B y 10% en células T. Se manifiesta por grandes adenopatías generalizadas e indoloras. Los síntomas B aparecen en el 40% de los casos; pueden diseminarse a otras cadenas ganglionares, bazo y médula ósea. Las formas extraganglionares son frecuentes (hepática de un 25 a 50%). Los podemos clasificar en indolentes y agresivos. Dentro de éstos últimos vamos a referirnos específicamente al Linfoma T periférico.
El linfoma T periférico afecta a adultos, aunque también a niños. Es una enfermedad diseminada, que ocasionalmente cursa con eosinofilia, prurito o síndrome hemofagocítico. Puede afectar ganglios linfáticos, piel, hígado, bazo y otras vísceras. El curso es agresivo pero potencialmente curable. Esta podría ser una variante posible para el diagnóstico de nuestro paciente. En contra del mismo es que no presenta eosinofilia ni prurito.
La variante linfoma anaplásico de células grandes tipo T es un linfoma sistémico de células T que se caracteriza por afectar a adultos jóvenes y niños (3% de linfomas no Hodgkin del adulto y 25-30% en niños) y habitualmente a varones. Clínicamente son muy agresivos, los síntomas B son frecuentes y tiene frecuente afectación extraganglionar, alrededor del 60 % (piel y médula ósea 5-30%). Las características clínicas coinciden con las del paciente por lo cual considero a esta la variante más probable. El linfoma difuso de células grandes tipo B representa el 30 % de los LNH. Es más frecuente en varones y mayores de 60 años. Se presentan como una masa única de crecimiento rápido. La mitad tiene afectación extranodal siendo más frecuente el compromiso del SNC, hueso y aparato gastrointestinal. Es rara la afectación de médula ósea. En este caso el paciente era joven, presentaba poliadenopatías y no una masa única de crecimiento rápido; además el compromiso medular es claro por lo que este diagnóstico parece muy poco probable.
La variante linfoma anaplásico de células grandes tipo T es un linfoma sistémico de células T que se caracteriza por afectar a adultos jóvenes y niños (3% de linfomas no Hodgkin del adulto y 25-30% en niños) y habitualmente a varones. Clínicamente son muy agresivos, los síntomas B son frecuentes y tiene frecuente afectación extraganglionar, alrededor del 60 % (piel y médula ósea 5-30%). Las características clínicas coinciden con las del paciente por lo cual considero a esta la variante más probable. El linfoma difuso de células grandes tipo B representa el 30 % de los LNH. Es más frecuente en varones y mayores de 60 años. Se presentan como una masa única de crecimiento rápido. La mitad tiene afectación extranodal siendo más frecuente el compromiso del SNC, hueso y aparato gastrointestinal. Es rara la afectación de médula ósea. En este caso el paciente era joven, presentaba poliadenopatías y no una masa única de crecimiento rápido; además el compromiso medular es claro por lo que este diagnóstico parece muy poco probable.
Las Mastocitosis engloban un conjunto de enfermedades caracterizadas por un acúmulo de mastocitos en la piel, con o sin afectación de otros órganos o sistemas. Las diferentes formas clínicas se clasifican como:
1. Cutánea:-Urticaria pigmentosa
-Mastocitoma solitario
-Telangiectasia macularis eruptiva perstans
- Mastocitosis cutánea difusa
-Mastocitoma solitario
-Telangiectasia macularis eruptiva perstans
- Mastocitosis cutánea difusa
2. Mastocitosis sistémica
3. Maligna (leucemia mastocítica)
Las formas cutáneas son generalmente propias de la infancia y se pueden acompañar de manifestaciones clínicas generalizadas con o sin infiltración sistémica. Haré referencia especialmente a la mastocitosis sistémica la cual se caracteriza por presentar un incremento patológico de mastocitos pero en otros tejidos distintos al cutáneo.
La mastocitosis sistémica es más frecuente en adolescentes y adultos. Los síntomas sistémicos pueden ser inespecíficos (astenia, anorexia, pérdida de peso) o específicos debidos a la liberación de los mediadores inflamatorios (crisis de flushing, cefalea, diarrea). La afectación más frecuente es la ósea (70%) y la hepatoesplenomegalia (50 %) secundaria a infiltración mastocitaria. Se puede afectar el aparato gastrointestinal, con casos de úlcera péptica, malaabsorción por atrofia parcial de las vellosidades intestinales que da lugar a diarrea, dolor abdominal y vómitos. Puede haber infiltración de la médula ósea asociada a una eosinofília periférica, anemia y leucocitosis (en un 33% de los pacientes); como así también de los ganglios linfáticos, con adenopatías generalizadas.
La mastocitosis maligna es la transformación maligna de los mastocitos, denominada también leucemia mastocítica. Esta parece ser un proceso raro, aunque no excepcional. La sintomatología es muy similar a la mastocitosis sistémica pero con un curso más agresivo y fulminante.
Con respecto a nuestro paciente no presentaba compromiso óseo que es muy frecuente en esta enfermedad, no tenia eosinofilia periférica, afectación gastrointestinal ni los síntomas derivados de la liberación de los mediadores inflamatorios de los mastocitos.
La mastocitosis sistémica es más frecuente en adolescentes y adultos. Los síntomas sistémicos pueden ser inespecíficos (astenia, anorexia, pérdida de peso) o específicos debidos a la liberación de los mediadores inflamatorios (crisis de flushing, cefalea, diarrea). La afectación más frecuente es la ósea (70%) y la hepatoesplenomegalia (50 %) secundaria a infiltración mastocitaria. Se puede afectar el aparato gastrointestinal, con casos de úlcera péptica, malaabsorción por atrofia parcial de las vellosidades intestinales que da lugar a diarrea, dolor abdominal y vómitos. Puede haber infiltración de la médula ósea asociada a una eosinofília periférica, anemia y leucocitosis (en un 33% de los pacientes); como así también de los ganglios linfáticos, con adenopatías generalizadas.
La mastocitosis maligna es la transformación maligna de los mastocitos, denominada también leucemia mastocítica. Esta parece ser un proceso raro, aunque no excepcional. La sintomatología es muy similar a la mastocitosis sistémica pero con un curso más agresivo y fulminante.
Con respecto a nuestro paciente no presentaba compromiso óseo que es muy frecuente en esta enfermedad, no tenia eosinofilia periférica, afectación gastrointestinal ni los síntomas derivados de la liberación de los mediadores inflamatorios de los mastocitos.
La enfermedad maligna de los histiocitos son proliferaciones malignas de las células terminales de la diferenciación monocítica, es decir, histiocitos y células dendríticas. Son muy raros y se observan en adolescentes. La médula ósea es el órgano que se infiltra con más frecuencia; también el bazo, el hígado y los ganglios linfáticos. Su curso es rápidamente progresivo, a veces fulminante. Clínicamente se puede manifestar con fiebre y mal estado general, pérdida de peso, debilidad y sudoración. Cursan con hepatoesplenomegalia, adenopatías y lesiones cutáneas. Es raro observar infiltrados pulmonares, lesiones óseas y afección del SNC. En sangre periférica suele observarse pancitopenia secundaria a la infiltración histiocítica de la médula ósea, y se pueden detectar células histiocíticas atípicas circulantes. La evolución de la enfermedad es rápidamente progresiva y, sin tratamiento, el enfermo fallece en un plazo de 2-6 meses. Es importante realizar diagnóstico diferencial con las histiocitosis reactivas, sobre todo aquellas secundarias a virus y enfermedad de Hodgkin.
Las leucemias agudas son proliferaciones neoplásicas de progenitores inmaduros de la médula ósea. Mencionaré especialmente la leucemia linfoblástica aguda la cual representa el 20 % de las neoplasias en los adultos. Puede manifestarse con leucopenia o leucocitosis pudiendo infiltrar hígado, bazo, testículos, pero principalmente el SNC y pueden detectarse adenopatías periféricas. No parece ser el caso de nuestro paciente debido a que el mismo no presenta compromiso ni del SNC ni infiltración testicular.
Concluyendo en relación a la enfermedad de base, considero que probablemente este paciente haya tenido una enfermedad linfoproliferativa, particularmente el linfoma T periférico variante anaplásico de células grandes ya que suele afectar a varones adultos jóvenes y tiene frecuente afectación extraganglionar, sobre todo medula ósea lo que explicaría la pancitopenia. La ausencia de síntomas B quizás este en relación a que estaba en tratamiento con corticoides por las lesiones cutáneas de las cuales voy a hablar a continuación.
¿Las lesiones en miembros inferiores que presentaba el paciente pudieron estar relacionadas con su enfermedad de base?
Concluyendo en relación a la enfermedad de base, considero que probablemente este paciente haya tenido una enfermedad linfoproliferativa, particularmente el linfoma T periférico variante anaplásico de células grandes ya que suele afectar a varones adultos jóvenes y tiene frecuente afectación extraganglionar, sobre todo medula ósea lo que explicaría la pancitopenia. La ausencia de síntomas B quizás este en relación a que estaba en tratamiento con corticoides por las lesiones cutáneas de las cuales voy a hablar a continuación.
¿Las lesiones en miembros inferiores que presentaba el paciente pudieron estar relacionadas con su enfermedad de base?
Existen varias dermatosis paraneoplásicas que son poco frecuentes y de difícil reconocimiento. Se presentan predominantemente en adultos jóvenes entre la tercera y cuarta décadas de la vida y están asociadas con mayor frecuencia a enfermedades linfoproliferativas. Pueden aparecer antes o simultáneamente con la neoplasia, siendo raro que aparezca posteriormente.
Se reconocen las siguientes entidades: acantosis nigricans, acroqueratosis paraneoplásica (síndrome de Bazex), dermatomiositis, síndrome de Sweet, eritema necrolítico migratorio, hipertricosis lanuginosa, pénfigo paraneoplásico, eritema gyratum repens, queratosis seborreica eruptiva (signo de Lesser – Trélat) y pioderma gangrenoso. Voy a comentar este último por frecuencia y debido a que las lesiones tenían estas características.
El pioderma gangrenoso se caracteriza clínicamente por la presencia de úlceras irregulares, de bordes socavados, base necrótica y purulenta, perforaciones que drenan pus y escara necrótica. Estas lesiones se localizan más frecuentemente en miembros inferiores y también pueden afectar glúteos, abdomen y cara. Se asocian a trastornos mielo y linfoproliferativos.
Por otra parte las vasculitis paraneoplásicas pueden encontrarse como manifestación de una enfermedad linfoproliferativa. Este término se refiere a la inflamación y necrosis de las paredes vasculares cuya expresión clínica es variable y que se caracteriza por la afectación frecuente de piel y tejido celular subcutáneo. Pueden ser marcadores de la presencia de un cáncer visceral y es importante su reconocimiento debido a que facilita el diagnóstico precoz de un cáncer oculto. En los pacientes con vasculitis la asociación con una neoplasia maligna tiene una prevalencia media del 5 %. La vasculitis leucocitoclástica es la forma más frecuente y en segundo lugar la panarteritis nodosa.
La aparición de la vasculitis puede ser previa, simultánea o posterior a la neoplasia. La incidencia es claramente mayor en relación con neoplasias hematológicas que con tumores sólidos. Se ha destacado la asociación con neoplasias linfoideas, en especial síndromes linfoproliferativos (como la tricoleucemia hasta un 20 %) y síndromes mielodisplásicos (3 a 5 %). Se debe sospechar la presencia de una vasculitis paraneoplásica en todos los casos en los que el paciente no presente condicionantes definidos para el desarrollo de vasculitis o la falta de respuesta al tratamiento.
El granuloma de Churg Strauss, hallazgo histológico de este paciente en la biopsia cutánea se caracteriza por presentar necrosis basofílica central en la dermis media y degeneración fibrilar del colágeno, proliferación de histiocitos en empalizada rodeando el área necrótica; infiltrados de eosinófilos y neutrófilos (en 30 a 40% de los casos). Recibe varias denominaciones: dermatitis granulomatosa intersticial, granuloma necrosante extravascular cutáneo, pápulas reumatoides y dermatitis granulomatosa neutrofílica en empalizada. El hallazgo del granuloma extravascular cutáneo necrosante debe ser considerado un indicador de la presencia de una enfermedad inmunorreactiva concomitante. Las manifestaciones cutáneas pueden ser maculopápulas eritematosas similares al eritema multiforme, lesiones hemorrágicas, nódulos cutáneos y en tejido celular subcutáneo, úlceras, livedo reticularis y edema facial. El granuloma de Churg Strauss y sus manifestaciones clínicas pueden comportarse como lesiones paraneoplásicas.
Aparecen en asociación con linfomas en dos situaciones:
El pioderma gangrenoso se caracteriza clínicamente por la presencia de úlceras irregulares, de bordes socavados, base necrótica y purulenta, perforaciones que drenan pus y escara necrótica. Estas lesiones se localizan más frecuentemente en miembros inferiores y también pueden afectar glúteos, abdomen y cara. Se asocian a trastornos mielo y linfoproliferativos.
Por otra parte las vasculitis paraneoplásicas pueden encontrarse como manifestación de una enfermedad linfoproliferativa. Este término se refiere a la inflamación y necrosis de las paredes vasculares cuya expresión clínica es variable y que se caracteriza por la afectación frecuente de piel y tejido celular subcutáneo. Pueden ser marcadores de la presencia de un cáncer visceral y es importante su reconocimiento debido a que facilita el diagnóstico precoz de un cáncer oculto. En los pacientes con vasculitis la asociación con una neoplasia maligna tiene una prevalencia media del 5 %. La vasculitis leucocitoclástica es la forma más frecuente y en segundo lugar la panarteritis nodosa.
La aparición de la vasculitis puede ser previa, simultánea o posterior a la neoplasia. La incidencia es claramente mayor en relación con neoplasias hematológicas que con tumores sólidos. Se ha destacado la asociación con neoplasias linfoideas, en especial síndromes linfoproliferativos (como la tricoleucemia hasta un 20 %) y síndromes mielodisplásicos (3 a 5 %). Se debe sospechar la presencia de una vasculitis paraneoplásica en todos los casos en los que el paciente no presente condicionantes definidos para el desarrollo de vasculitis o la falta de respuesta al tratamiento.
El granuloma de Churg Strauss, hallazgo histológico de este paciente en la biopsia cutánea se caracteriza por presentar necrosis basofílica central en la dermis media y degeneración fibrilar del colágeno, proliferación de histiocitos en empalizada rodeando el área necrótica; infiltrados de eosinófilos y neutrófilos (en 30 a 40% de los casos). Recibe varias denominaciones: dermatitis granulomatosa intersticial, granuloma necrosante extravascular cutáneo, pápulas reumatoides y dermatitis granulomatosa neutrofílica en empalizada. El hallazgo del granuloma extravascular cutáneo necrosante debe ser considerado un indicador de la presencia de una enfermedad inmunorreactiva concomitante. Las manifestaciones cutáneas pueden ser maculopápulas eritematosas similares al eritema multiforme, lesiones hemorrágicas, nódulos cutáneos y en tejido celular subcutáneo, úlceras, livedo reticularis y edema facial. El granuloma de Churg Strauss y sus manifestaciones clínicas pueden comportarse como lesiones paraneoplásicas.
Aparecen en asociación con linfomas en dos situaciones:
· Con compromiso de la piel por una variante granulomatosa de un linfoma de células T.
· Puede detectarse en la piel de pacientes con linfomas sistémicos sin evidencia de afectación cutánea por el linfoma.
· Puede detectarse en la piel de pacientes con linfomas sistémicos sin evidencia de afectación cutánea por el linfoma.
Si bien esta asociación es rara y hay pocos casos reportados, podría haber sido la de nuestro paciente.
¿Cuál fue la causa de muerte de este paciente?
¿Cuál fue la causa de muerte de este paciente?
Si asociamos la falla multiorgánica, la taquicardia, la taquipnea, la leucopenia y el hallazgo del desarrollo de Pseudomona aeruginosa en sangre podemos afirmar que este paciente presentó un cuadro de sepsis. Posteriormente estando en terapia intensiva agrega hipotensión y acidosis metabólica evolucionando a shock séptico, el cual creo que es el que produce la muerte. La presencia de una P. aeruginosa en sangre define la sepsis como causa de la falla multiorgánica.
Sabemos que se llama sepsis a la presencia de síndrome de respuesta inflamatoria sistémica (fiebre mayor de 38º C o hipotermia menor de 36º C, frecuencia cardiaca superior a 90 lpm, taquipnea con más de 20 cpm o paCO2 menor de 32 mmHg, alteración del recuento de leucocitos con más de 12.000 o menos de 4.000 leucocitos/mm3, o más del 10% de formas en cayado) secundario a infección documentada. Se denomina sepsis grave cuando se asocia a hipotensión y shock séptico hipotensión que persiste a pesar de la administración de fluidos, acompañada de alteraciones de la perfusión (acidosis metabólica, hiperlactacidemia) o disfunción de órganos o necesidad de fármacos vasoactivos para mantener la presión arterial.
La Pseudomona Aeruginosa, es un gérmen que se encuentra ampliamente difundido en la naturaleza, constituyendo su hábitat el agua, las plantas, la tierra húmeda, las colecciones artificiales de agua, como piscinas, depósitos, incluso se la llega a aislar en líquidos "desinfectantes”. Los factores de riesgo para infección por P. Aeruginosa son enfermedad pulmonar estructural (bronquiectasias), terapéutica prolongada con corticosteroides (más de 10 mg diarios de prednisona), terapia con antibióticos de amplio espectro el mes anterior durante más de 7 días, desnutrición, infección por el virus de la inmunodeficiencia humana (VIH) y neutropenia. Clínicamente puede provocar: bacteriemia, neumonía, endocarditis, osteomielitis y artritis, meningitis, infecciones urinarias, infección de partes blandas, infecciones oftálmicas y otorrinolaringológicas e infecciones del tubo digestivo. El compromiso pulmonar en las formas bacteriémicas suele aparecer en pacientes neutropénicos y tiene una extraordinaria gravedad. La anatomía patológica muestra necrosis de las paredes alveolares, hemorragia y formación de microabscesos. Representa un 10%-20% del total de sepsis por gérmenes Gram negativos y tiene una alta mortalidad (40%-70%). Las puertas de entrada son muy diversas: quemaduras (piel), tubo digestivo, pulmón, vías urinarias y catéteres intravenosos.
Los factores de mal pronóstico ante una bacteriemia por P. aeruginosa son: neutropenia persistente o inferior a 100 células/mm3, insuficiencia renal, focos metastásicos, shock séptico, presencia de enfermedad fatal, terapia antibiótica inadecuada y origen pulmonar, cutáneo o desconocido.
Teniendo en cuenta todos estos datos es lógico pensar que este paciente con múltiples factores de riesgo para el desarrollo de una infección por P aeruginosa (probable neoplasia hematológica de base, tratamiento con corticoides previo, etc.) haya presentado una sepsis grave que produjo el deceso.
Conclusión:
Sabemos que se llama sepsis a la presencia de síndrome de respuesta inflamatoria sistémica (fiebre mayor de 38º C o hipotermia menor de 36º C, frecuencia cardiaca superior a 90 lpm, taquipnea con más de 20 cpm o paCO2 menor de 32 mmHg, alteración del recuento de leucocitos con más de 12.000 o menos de 4.000 leucocitos/mm3, o más del 10% de formas en cayado) secundario a infección documentada. Se denomina sepsis grave cuando se asocia a hipotensión y shock séptico hipotensión que persiste a pesar de la administración de fluidos, acompañada de alteraciones de la perfusión (acidosis metabólica, hiperlactacidemia) o disfunción de órganos o necesidad de fármacos vasoactivos para mantener la presión arterial.
La Pseudomona Aeruginosa, es un gérmen que se encuentra ampliamente difundido en la naturaleza, constituyendo su hábitat el agua, las plantas, la tierra húmeda, las colecciones artificiales de agua, como piscinas, depósitos, incluso se la llega a aislar en líquidos "desinfectantes”. Los factores de riesgo para infección por P. Aeruginosa son enfermedad pulmonar estructural (bronquiectasias), terapéutica prolongada con corticosteroides (más de 10 mg diarios de prednisona), terapia con antibióticos de amplio espectro el mes anterior durante más de 7 días, desnutrición, infección por el virus de la inmunodeficiencia humana (VIH) y neutropenia. Clínicamente puede provocar: bacteriemia, neumonía, endocarditis, osteomielitis y artritis, meningitis, infecciones urinarias, infección de partes blandas, infecciones oftálmicas y otorrinolaringológicas e infecciones del tubo digestivo. El compromiso pulmonar en las formas bacteriémicas suele aparecer en pacientes neutropénicos y tiene una extraordinaria gravedad. La anatomía patológica muestra necrosis de las paredes alveolares, hemorragia y formación de microabscesos. Representa un 10%-20% del total de sepsis por gérmenes Gram negativos y tiene una alta mortalidad (40%-70%). Las puertas de entrada son muy diversas: quemaduras (piel), tubo digestivo, pulmón, vías urinarias y catéteres intravenosos.
Los factores de mal pronóstico ante una bacteriemia por P. aeruginosa son: neutropenia persistente o inferior a 100 células/mm3, insuficiencia renal, focos metastásicos, shock séptico, presencia de enfermedad fatal, terapia antibiótica inadecuada y origen pulmonar, cutáneo o desconocido.
Teniendo en cuenta todos estos datos es lógico pensar que este paciente con múltiples factores de riesgo para el desarrollo de una infección por P aeruginosa (probable neoplasia hematológica de base, tratamiento con corticoides previo, etc.) haya presentado una sepsis grave que produjo el deceso.
Conclusión:
· Enfermedad de base: Linfoma de alto grado probablemente tipo T, variante anaplásica.
· Lesiones dermatológicas con granuloma de Churg Strauss pueden ser paraneoplásicas secundarias a un linfoma de afectación sistémica.
· Causa de muerte: sepsis por P. aeruginosa.
· Lesiones dermatológicas con granuloma de Churg Strauss pueden ser paraneoplásicas secundarias a un linfoma de afectación sistémica.
· Causa de muerte: sepsis por P. aeruginosa.
Diagnóstico anatomopatológico:
Anatomía Patológica: Dr. Mario Gorosito (Cátedra Anatomía Patológica UNR)
Se presentó el caso de un paciente de sexo masculino de 38 años que presentaba un cuadro de un año de evolución caracterizado por placas eritematosas, edematosas, infiltradas, dolorosas sobre las que se agregan ampollas que desecan y forman costras necróticas, algunas de las cuales curan dejando cicatriz. Se realizó una biopsia de piel de miembro inferior cuyas características morfológicas corresponderían a un patrón de reacción tipogranuloma de Churg Strauss.
En su momento se comentó la necesidad de correlacionar estos hallazgos con la clínica, la evolución y el laboratorio del paciente.
En 1983 se describe por primera vez esta entidad con el término granuloma de Churg Strauss o granuloma cutáneo extravascular necrosante (Winkelmann granuloma). Otros autores describieron lesiones o patrones similares utilizando términos como: necrobiosis reumatoide superficial ulcerativa (pápulas reumatoideas); dermatitis granulomatosa intersticial con nódulos cutáneos y artritis; dermatitis intersticial granulomatosa con placas entre otros.
En realidad, se trata de un patrón histopatológico asociado a enfermedades sistémicas autoinmunes siendo su etiología desconocida, pero se sospecha sea de índole inmunorreactiva, ya que se presenta en pacientes con diversos desórdenes inmunológicos, asociado a autoanticuerpos y formación de inmunocomplejos.
La manifestación cutánea puede preceder a la manifestación clínica sistémica de la enfermedad o desarrollarse durante ésta o incluso después.
Las patologías más frecuentemente asociadas a éste patrón histopatológicos son:
En 1983 se describe por primera vez esta entidad con el término granuloma de Churg Strauss o granuloma cutáneo extravascular necrosante (Winkelmann granuloma). Otros autores describieron lesiones o patrones similares utilizando términos como: necrobiosis reumatoide superficial ulcerativa (pápulas reumatoideas); dermatitis granulomatosa intersticial con nódulos cutáneos y artritis; dermatitis intersticial granulomatosa con placas entre otros.
En realidad, se trata de un patrón histopatológico asociado a enfermedades sistémicas autoinmunes siendo su etiología desconocida, pero se sospecha sea de índole inmunorreactiva, ya que se presenta en pacientes con diversos desórdenes inmunológicos, asociado a autoanticuerpos y formación de inmunocomplejos.
La manifestación cutánea puede preceder a la manifestación clínica sistémica de la enfermedad o desarrollarse durante ésta o incluso después.
Las patologías más frecuentemente asociadas a éste patrón histopatológicos son:
Enfermedades linfoproliferativas, tiroiditis autoinmune, lupus eritematoso sistémico, vasculitis sistémicas, granulomatosis de Wegener, tiroiditis autoinmune, enfermedad inflamatoria intestinal y artritis reumatoidea.
El diagnóstico de la punción biopsia de médula ósea fue infiltración medular por una proliferación celular de mediano y gran tamaño, sugestiva de corresponder a un proceso linfoproliferativo (Linfoma No Hodgkin de alto grado). Se considera necesario realizar estudios inmunohistoquímicos.
El diagnóstico de la punción biopsia de médula ósea fue infiltración medular por una proliferación celular de mediano y gran tamaño, sugestiva de corresponder a un proceso linfoproliferativo (Linfoma No Hodgkin de alto grado). Se considera necesario realizar estudios inmunohistoquímicos.

Otras punciones biopsia postmortem:
Punción de Hígado:
· Pequeños fragmentos de tejido hepático, sin espacios portales.
· Hay colestasis intrahepática de grado leve y de distribución difusa.
· Se observa esteatosis leve no sistematizada.
· No se observa lesión neoplásica ni inflamatoria.
· Pequeños fragmentos de tejido hepático, sin espacios portales.
· Hay colestasis intrahepática de grado leve y de distribución difusa.
· Se observa esteatosis leve no sistematizada.
· No se observa lesión neoplásica ni inflamatoria.
Punción de ganglio:
· Pequeños fragmentos de músculo estriado.
Estudio inmunohistoquímico de médula ósea
- Antígeno común leucocitario (ACL): positivo
-CD 20 (L26 ): negativo
-CD 3:positivo
-CD 30 (BER H2: positivo
-CD 15 (LEU M1): negativo
-ALK1: negativo
- Antígeno común leucocitario (ACL): positivo
-CD 20 (L26 ): negativo
-CD 3:positivo
-CD 30 (BER H2: positivo
-CD 15 (LEU M1): negativo
-ALK1: negativo


Comentarios: el inmunofenotipo, en correlación con la morfología, corresponde al compromiso medular por un linfoma anaplásico T, CD 30 positivo (Informe Dra. Sandra Sarancone).
Como comentario final, el Linfoma T anaplásico de células grandes es una variante de linfoma No Hodgkin de alto grado muy agresivos, CD 30+ y CD20+ en el estudio inmunohistoquímico, que se presenta en niños y adultos jóvenes habitualmente en estadios avanzados. Comprometen sitios nodales (masas mediastinales) y extranodales como piel, hueso, partes blandas, pulmón, hígado y menos frecuente, el sistema nervioso central. La infiltración a médula ósea es excepcional.
Cuando se presenta en piel como forma autolimitada similar a la Papulosis Linfomatoide, son de buen pronóstico. Este paciente presentó una manifestación paraneoplásica en piel tipo granuloma de Churg-Strauss y posteriormente se presentó con compromiso de médula ósea. Debido a que no contamos con la necropsia, no podemos saber la extensión real del proceso ni la causa de muerte.
Como comentario final, el Linfoma T anaplásico de células grandes es una variante de linfoma No Hodgkin de alto grado muy agresivos, CD 30+ y CD20+ en el estudio inmunohistoquímico, que se presenta en niños y adultos jóvenes habitualmente en estadios avanzados. Comprometen sitios nodales (masas mediastinales) y extranodales como piel, hueso, partes blandas, pulmón, hígado y menos frecuente, el sistema nervioso central. La infiltración a médula ósea es excepcional.
Cuando se presenta en piel como forma autolimitada similar a la Papulosis Linfomatoide, son de buen pronóstico. Este paciente presentó una manifestación paraneoplásica en piel tipo granuloma de Churg-Strauss y posteriormente se presentó con compromiso de médula ósea. Debido a que no contamos con la necropsia, no podemos saber la extensión real del proceso ni la causa de muerte.
Referencias:
1) Vega, F. “Linfoma Anaplásico de Célula Grande”. Department of hematopathology. Anderson Cancer Center. Houston, Texas.
2) Rolando, N; Wade, J; Davalos, M; et al. “The systemic inflammatory response syndrome in acute liver failure”. Hepatology 2000; 32:734-739.
3) Gellrich, J; Hakenberg, OW; Nauman, R; et al. “Primary renal non-hodgkin`s lymphoma”. Onkologie 2002; 25 (3): 273-277.
4) Dıez-Porres, L; Ríos-Blanco, J; Robles-Marhuenda, a; et al. “ANCA-associated vasculitis as paraneoplastic syndrome with colon cancer: a case report”. Lupus 2005; 14:632–634.
5) Hatzis, G ; Papachristodoulou, A ; Delladetsima, I ; et al. “Polyarteritis nodosa associated with cholangiocarcinoma”. Lupus 1998; 7: 301-306
6) Jacobsen, E. “Anaplastic Large-Cell Lymphoma, T-/Null-Cell Type”. Oncologist 2006; 11:831-840.
7) Brown, J; Skarin, A. “Clinical mimics of lymphoma”. Oncologist 2004; 9:406-416.
8) Longley, J; Duffy, TP; Kohn, S. “The mast cell and mast cell disease”. Journal of the American Academy of Dermatology 1995, 32 (4): 545-61; 562-4.
9) Hotchkiss, R; Karl, I . “Medical Progress: The Pathophysiology and Treatment of Sepsis”. N Engl J Med 2003; 348:138-150.
10) Dellon, E; Morris, S; Tang, W; et al. “Acute liver failure due to natural killer-like T-cell leukemia/lymphoma: A case report and review Literature”. Word Journal of Gastroenterology. 2006; 12(25): 4089-4092.
11) Calonje, J; Greaves, M. “Cutaneous extravascular necrotizing granuloma (Churg Strauss) as a paraneoplastic manifestation of non-Hodgkin Lymphoma”. Journal of the Royal Society of Medicine. 1993; 86.
12) Diette, KM; Caro, WA; Roenigk, HH. “Malignant lymphoma presenting witrh cutaneous granulomas”. J Am Acad. Dermatology 1984; 10: 896-902.
1) Vega, F. “Linfoma Anaplásico de Célula Grande”. Department of hematopathology. Anderson Cancer Center. Houston, Texas.
2) Rolando, N; Wade, J; Davalos, M; et al. “The systemic inflammatory response syndrome in acute liver failure”. Hepatology 2000; 32:734-739.
3) Gellrich, J; Hakenberg, OW; Nauman, R; et al. “Primary renal non-hodgkin`s lymphoma”. Onkologie 2002; 25 (3): 273-277.
4) Dıez-Porres, L; Ríos-Blanco, J; Robles-Marhuenda, a; et al. “ANCA-associated vasculitis as paraneoplastic syndrome with colon cancer: a case report”. Lupus 2005; 14:632–634.
5) Hatzis, G ; Papachristodoulou, A ; Delladetsima, I ; et al. “Polyarteritis nodosa associated with cholangiocarcinoma”. Lupus 1998; 7: 301-306
6) Jacobsen, E. “Anaplastic Large-Cell Lymphoma, T-/Null-Cell Type”. Oncologist 2006; 11:831-840.
7) Brown, J; Skarin, A. “Clinical mimics of lymphoma”. Oncologist 2004; 9:406-416.
8) Longley, J; Duffy, TP; Kohn, S. “The mast cell and mast cell disease”. Journal of the American Academy of Dermatology 1995, 32 (4): 545-61; 562-4.
9) Hotchkiss, R; Karl, I . “Medical Progress: The Pathophysiology and Treatment of Sepsis”. N Engl J Med 2003; 348:138-150.
10) Dellon, E; Morris, S; Tang, W; et al. “Acute liver failure due to natural killer-like T-cell leukemia/lymphoma: A case report and review Literature”. Word Journal of Gastroenterology. 2006; 12(25): 4089-4092.
11) Calonje, J; Greaves, M. “Cutaneous extravascular necrotizing granuloma (Churg Strauss) as a paraneoplastic manifestation of non-Hodgkin Lymphoma”. Journal of the Royal Society of Medicine. 1993; 86.
12) Diette, KM; Caro, WA; Roenigk, HH. “Malignant lymphoma presenting witrh cutaneous granulomas”. J Am Acad. Dermatology 1984; 10: 896-902.