Linfoma linfoblástico. A propósito de un caso
Los linfomas no Hodgkin constituyen un grupo heterogéneo de neoplasias malignas linfoproliferativas que tienen diferentes modelos de comportamiento y respuestas al tratamiento. Es mucho menos previsible que el linfoma de Hodgkin y tiene predilección mucho mayor por diseminarse hasta sitios extraganglionares. El pronóstico depende del tipo histológico, el estadio y el tratamiento.
Autores:
- Dra. Reina Genellys Fernández Camps. Especialista de Segundo Grado de Medicina Interna. Profesor Auxiliar de Medicina Interna. Consultante del Georgetown Public Hospital Corporation. Guyana.
- Hughley Hanoman. Médico General del Georgetown Public Hospital Corporation. Guyana
- Taishan Shanks. Médico General del Georgetown Public Hospital Corporation. Guyana
- Aneisha Udelle Moore. Médico General del Georgetown Public Hospital Corporation. Guyana
Caso Clínico
Nombre: OG
Edad: 19 años
Color de la piel: Negra.
Ocupación: Trabajador por cuenta propia
Nacionalidad: Guyanés.
Fecha de Ingreso: 13/2/2014
Fecha de Egreso: 7/3/2014
MI: Dolores generalizados del cuerpo y sudoraciones frías
HEA: Paciente con antecedentes de aparente buena salud el cual refiere hace ± 6 días comenzó a presentar dolores generalizados en todo el cuerpo sin precisar otras características así como sudoraciones frías nocturnas ocasionales, y mareos sin precisar tampoco otras características. Todo esto asociado con pérdida del apetito y pérdida de peso de aproximadamente 20 libras en las últimas 2 semanas. Niega fiebre constatada termométricamente, aunque refiere “sentirse caliente por dentro”. Con este cuadro acude al cuerpo de guardia del Georgetown Public Hospital Corporation y se decide su ingreso.
APP: No refiere
APF: No refiere
Hábitos tóxicos: alcohol ocasional.
Uso de medicamentos: multivitaminas, vitaminas del complejo B, ácido fólico y panadol
Operaciones: No refiere
Examen físico:
Mucosas normocoloreadas y húmedas.
No edemas.
Sistema Respiratorio: Murmullo vesicular normal, no estertores.
Frecuencia respiratoria: 18 respiraciones por minuto
Sistema cardiovascular: Ruidos cardiacos audibles y rítmicos, no soplos.
Frecuencia cardiaca: 80 latidos por minuto. Tensión arterial: 110/80 mmHg
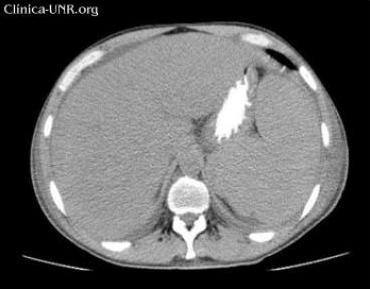
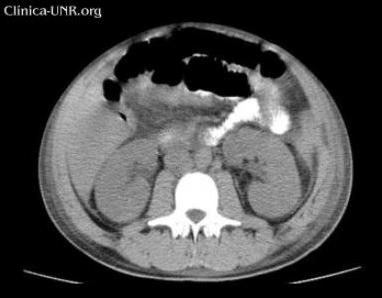
Abdomen: plano, depresible, doloroso a nivel de hipocondrio izquierdo donde se palpa aumento de volumen que parece corresponderse con esplenomegalia de ± 3cm, no dolorosa, de consistencia firme, bordes bien definidos y superficie lisa. Matidez a nivel de la celda esplénica. También se palpa hepatomegalia de ± 3cm superficial lisa, no dolorosa y bordes finos.
Sistema nervioso central (SNC): Consciente, orientado en tiempo, espacio y persona, no déficit motor. No signos meníngeos.
SHLP: se constatan adenomegalias de diferentes tamaño, la mayor de ± 3cm de diámetro, no dolorosas en todas las cadenas ganglionares del cuello, que no confluyen, bien delimitadas, no adheridas a planos profundos, movibles, sin cambios de la piel que las recubre, además de las cadenas submaxilar, axilar e inguinal.
Piel: petequias localizadas en ambos miembros inferiores.
Complementarios en el cuerpo de guardia:
Hemograma: Hb: 13,3g/dL
Leucocitos: 234,500/mm3
Poli: 0,3%
Linfo: 88%
Blastos: 0,5%
Prolinfocitos: 0,4%
Proliferación de la línea linfoide
Eritrosedimentación: 10mm/h
Plaquetas: 70,000/mm3
CK – MB: 59U/L (< 160U/L)
LDH: 126U/L (80 – 285U/L)
Fosfatasa alcalina: 289U/L (35 – 123U/L)
GGT: 169U/L (9 – 54IU/L)
TGO: 114IU/L (5 – 34IU/L)
TGP: 77IU/L (4 – 36U/L)
Tiempo de coagulación: 2min (5 – 10min)
Tiempo de sangramiento: 3min (1 – 3min)
Ionograma: Na: 137,1mmol/L
K: 4,71mmol/L
Cl: 96,0mmol/L
Análisis de orina: negativo
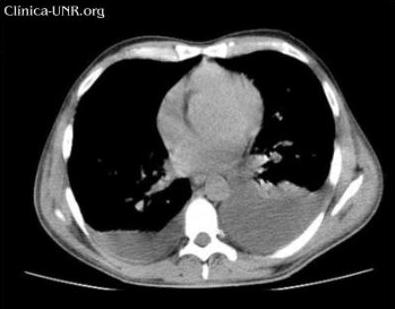
Rx de tórax: Ensanchamiento del mediastino por posible linfadenomegalias mediastinales.
ID: Leucemia Linfoblástica Aguda.
Tratamiento para sala de Medicina:
- Dieta libre
- SV cada 8 horas.
- Rocephin 1g IV diario.
- Panadol 500mg si fiebre.
- Omeprazol 20mg diarios VO.
- MV 1 tab diaria VO.
14/2/2014
Es visto el paciente por primera vez en sala por nuestro grupo básico de trabajo corroborándose los datos aportados por el paciente en la HC del cuerpo de guardia.
Al examen físico se corroboran las adenomegalias generalizadas con hepatoesplenomegalia.
Se indican complementarios:
- Repetir hemograma con diferencial y eritrosedimentación.
- Complementarios que midan la función hepática y renal.
- VIH (test rápido).
- US Abdominal.
- BAAF de ganglio.
15 – 17/2/2014
No se constatan cambios en la sintomatología de nuestro enfermo ni en el examen físico.
Se reciben complementarios:
Hemograma: Hb: 13,3g/dL
Leucocitos: 234,500/mm3
Poli: 0,1%
Linfo: 85%
Linfoblastos: 0,4%
Eritrosedimentación: 11mm/h
Plaquetas: 67,000/mm3
Acido urico: 2,5mg/dL (2,5 – 7,7mg/dL)
Calcio: 11,6mg/dL (8,5 – 10,4mg/dL)
LDH: 343U/L (80 – 285U/L)
Fosfatasa alcalina: 237U/L (35 – 123U/L)
Bilirrubina directa: 1,0mg/dL (0,0 – 0,5mg/dL)
GGT: 137U/L (9 – 54IU/L)
TGO: 105IU/L (5 – 34IU/L)
TGP: 72IU/L (4 – 36U/L)
Proteinas totals: 6,3g/dL (6,2 – 8,5g/dL)
Albumina: 3,5g/dL (3,5 – 5,3g/dL)
Globulina: 2,8g/dL
BUN: 47mg/dL (7 – 18mg/dL)
Creatinina: 1,7mg/dL (0,4 – 1,4mg/dL)
Ionograma: Na: 137,9mmol/L (135 – 148mmol/L)
K: 3,16mmol/L (3,5 – 5,3mmol/L)
Cl: 100,4mmol/L (98 – 107mmol/L)
Ultrasonido abdominal:
Ambos espacios pleurales claros.
Hígado aumentado de tamaño que excede 3cm el reborde costal, con textura homogénea. Conductos biliares intrahepáticos no dilatados.
Vesícula normal en tamaño, forma y posición. No cálculos
Esplenomegalia.
Ambos riñones normales en tamaño, forma y posición, con aumento de la ecogenicidad. Relación seno – parénquima conservada. No litiasis. No hidronefrosis.
No adenopatías visibles. Aorta abdominal normal.
No ascitis.
Vejiga normal en contorno y paredes engrosadas. No litiasis.
Se toma muestra para biopsia de ganglio.
18/2/2014
Mantiene síntomas y signos anteriormente descritos.
Complementarios recibidos:
Hemograma: Hb: 12,3g/dL
Leucocitos: 291,200/mm3
Poli: 0,6%
Linfo: 20%
Blastos: 74%
Eritrosedimentación: 12mm/h
Plaquetas: 118,000/mm3
PCV: 37%
BUN: 51mg/dL (7 – 18mg/dL)
Creatinina: 3,5mg/dL (0,4 – 1,4mg/dL)
Proteínas totales: 7,4g/dL (6,2 – 8,5g/dL)
Albúmina: 2,5g/dL (3,5 – 5,5g/dL)
Globulina: 4,9g/dL
Bilirrubina directa: 1,1mg/dL (0,0 – 0,5mg/dL)
Ionograma: Na: 134,5mmol/L (135 – 148mmol/L)
K: 5,16mmol/L (3,5 – 5,3mmol/L)
Cl: 95,4mmol/L (98 – 107mmol/L)
Se decide comenzar con albumina humana 500mg diarios IV.
19 – 20/2/2014
Mantiene sintomatología y hallazgos al examen físico.
BAAF de ganglio: Linfoma Linfoblástico.
Se realiza Medulograma
21/2/2014
Clínicamente estable, igual sintomatología y sin cambios en el examen físico.
Complementarios:
Hemograma: Hb: 7,0g/dL
Leucocitos: 240,000/mm
3 Linfoblastos: 0,9%
Plaquetas: 56,000/mm3
BUN: 40mg/dL (7 – 18mg/dL)
Creatinina: 1,7mg/dL (0,4 – 1,4mg/dL)
Acido úrico: 6,2mg/dL (2,5 – 7,7mg/dL)
Ionograma: Na: 129,7mmol/L (135 – 148mmol/L)
K: 4,14mmol/L (3,5 – 5,3mmol/L)
Cl: 92,7mmol/L (98 – 107mmol/L)
Aspiración de médula ósea:
Médula ósea hipercelular con gran proliferación de células linfoides
Diferencial: Linfoblastos: 23%
Linfocitos: 71%
Neutrofilos: 5%
Eosinofilos: 1%
Conclusiones: Linfoma no Hodgkin Linfoblástico
Se le impone tratamiento con hidratación de suero salino fisiológico (SSF) para corregir la hiponatremia (no se cuenta con cloro-sodio hipertónico en el hospital ni el país)
Fig. 1. Aspirado de Médula Ósea.
22 – 25/2/2014
Se mantiene estable y sin cambios en el examen físico.
Es valorado por Nefrología sin criterios en ese momento de tratamiento depurador renal y lo considera una Insuficiencia Renal Aguda secundaria a Linfoma no Hodgkin.
Sugiere administrar líquidos según necesidades a 1500ml/m2sc/día y comenzar tratamiento con Alopurinol.
26 – 28/2/2014
Se mantiene estable clínicamente y es valorado en el Instituto del Cáncer de Guyana, concluyéndose como un Linfoma no Hodgkin Linfoblástico, decidiéndose comenzar ciclo de quimioterapia (se le administró el ciclo el dia 28/2/2014) por 6 ciclos con::
Premedicación:
- Ondansetron: 8mg IV
- Dexametazona: 4mg IV
- Ranitidina (Zantac) 50mg IV
Quimioterapia:
- Rituximab 670mg en 500cc de SSF IV a durar 2 horas.
- Ciclofosfamida 1335mg en 500cc de SSF IV a durar 2 horas.
- Doxorubicina: 90mg en 500cc de SSF IV a durar 2 horas.
- Vincristina: 2mg IV.
- Prednisona 70mg diarios por VO por 5 días.
- Una vez terminada la medicación administrar 500cc SSF en bolo a durar 4 horas.
Previa medicación con citostáticos se decide administrar 2 unidades de plaquetas por tener el mismo una plaquetopenia secundaria a su enfermedad de base.
1 – 2/3/2014
Se mantiene estable y es evolucionado por la guardia médica
Se indican complementarios evolutivos.
Se transfunde con 2 unidades de glóbulos.
3/3/2014
Refiere haber tenido 3 deposiciones diarreicas acuosas, no sangre, no flemas.
Complementarios:
Hemograma: Hb: 9,9g/dL
Leucocitos: 140,600/mm3
Plaquetas: 41,000/mm3
Albumina: 3,9g/dL
BUN: 71mg/dL
Creatinina: 2,7mg/dL
Tiempo de sangramiento: 3 min 15 seg
Tiempo de coagulación: 2 min 40 seg
4 – 5/3/2014
Se mantiene estable y sin cambios en el examen físico
Complementarios:
Hemograma: Hb: 9,0g/dL
Plaquetas: 32,000/mm3
BUN: 43mg/dL (7 – 18mg/dL)
Creatinina: 1,7mg/dL (0,4 – 1,4mg/dL)
Ionograma: Na: 135,0mmol/L (135 – 148mmol/L)
K: 4,55mmol/L (3,5 – 5,3mmol/L)
Cl: 100,9mmol/L (98 – 107mmol/L)
AgHBs: negativo.
Se mantiene tratamiento con antibioticoterapia (Augmentin 1,2g IV c/12 horas), protección gástrica (Omeprazol 20m 2 veces al dia), Alopurinol (50mg diarios) Prednisona 70mg diarios (como parte del tratamiento quimioterápico) y se transfunde nuevamente con glóbulos (1 unidad) y plaquetas (2 unidades).
6/3/2014
Estable y asintomático, sin cambios en el examen físico.
Complementarios
Hemograma: Hb: 9,37/dL
Leucocitos: 25,100/mm3
Poli: 0,9%
Linfo: 21,3%
Mono: 2,3%
Plaquetas: 39,000/mm3
PCV: 37%
Albúmina: 3,8g/dL (3,5 – 5,5g/dL)
GGT: 186U/L (9 – 54IU/L)
TGO:
43IU/L (5 – 34IU/L)
TGP: 44IU/L (4 – 36U/L)
Bilirrubina total: 1,2mg/dL (0,2 – 1,2mg/dL
Bilirrubina directa: 0,1mg/dL (0,0 – 0,5mg/dL)
Bilirrubina indirecta: 1,1mg/dL (0,1 – 1,1mg/dL)
Acido urico: 2,5mg/dL (2,5 – 7,7mg/dL)
BUN: 49mg/dL (7 – 18mg/dL)
Creatinina: 0,7mg/dL (0,4 – 1,4mg/dL)
Ionograma: Na: 132,9mmol/L (135 – 148mmol/L)
K: 4,56mmol/L (3,5 – 5,3mmol/L)
Cl: 100,9mmol/L (98 – 107mmol/L)
Tiempo de coagulación: 4 min (5 – 10min)
Tiempo de sangramiento: 3 min (1 – 3min)
Se decide egresar con seguimiento por consulta para continuar tratamiento oncoespecífico
Comentario.
Linfoma es el nombre de un grupo de tipos de cáncer de la sangre que comienzan en el sistema linfático. Alrededor del 54 por ciento de los casos de cáncer de la sangre que se presentan cada año son tipos de linfoma. El linfoma se presenta cuando un linfocito (un tipo de glóbulo blanco) sufre un cambio maligno y se multiplica; con el tiempo estas células malignas desplazan a las células sanas y crean tumores. Estos tumores causan un agrandamiento de los ganglios linfáticos y/o crecen en otros lugares que forman parte del sistema inmunitario (por ejemplo, la piel y otros órganos). La leucemia linfocítica, un tipo de cáncer de la sangre que también se origina en un linfocito, está estrechamente relacionada con el linfoma.
Existen dos tipos principales de linfoma: el linfoma de Hodgkin y el linfoma no Hodgkin (NHL, por sus siglas en inglés), existiendo de este último más de 60 subtipos.
Los linfomas no Hodgkin constituyen un grupo heterogéneo de neoplasias malignas linfoproliferativas que tienen diferentes modelos de comportamiento y respuestas al tratamiento. Es mucho menos previsible que el linfoma de Hodgkin y tiene predilección mucho mayor por diseminarse hasta sitios extraganglionares. El pronóstico depende del tipo histológico, el estadio y el tratamiento.
Los oncólogos además clasifican los subtipos de linfoma no Hodgkin (NHL) según la tasa de progresión de la enfermedad, o sea, si son agresivos (de progresión rápida) o indolentes (de progresión lenta). El subtipo de NHL (y si es de forma agresiva o indolente) determina el tratamiento adecuado, por lo que es muy importante obtener un diagnóstico correcto.
Cuadro 1. Clasificación según la tasa de progresión de le enfermedad.
Subtipos Agresivos: linfoma no Hodgkin (NHL) de progresión rápida o de alto grado
Linfoma asociado al SIDA*
Linfoma anaplásico de células grandes
Linfoma de Burkitt
*
Linfoma del Sistema Nervioso Central
Linfoma difuso de células B grandes (es el más común de los NHL agresivo)
Linfoma Linfoblástico*
Linfoma de células del manto
Linfoma periférico de células T (la mayoría de los subtipos)
Linfomas/leucemias de precursores de células B y T
Linfoma folicular transformado y Linfoma de tejido linfoide asociado a las mucosas (MALT, por sus siglas en ingles) transformado
*El linfoma de Burkitt, el linfoma asociado al SIDA, y el linfoma linfoblástico se clasifican como “subtipos muy agresivos”
Subtipos Indolentes: linfoma no Hodgkin (NHL) de progresión lenta o de bajo grado
Linfoma cutáneo de células T (Micosis fungoide y Síndrome de Sézary)
Linfoma folicular (es el más común de los NHL Indolentes)
Linfoma linfoplasmacítico y Macroglobulinemia de Wldeströn
Linfoma de tejido linfoide asociado a las mucosas (MALT)
Linfoma ganglionar de células B de la zona marginal
Linfoma linfocítico de células pequeñas/leucemia linfocítica crónica
Se considera que algunos pacientes tienen una enfermedad de “grado intermedio”, que tiene una tasa de progresión entre agresiva e indolente. Algunos casos de linfoma no Hodgkin (NHL) indolente se “transforman” en NHL agresivo.
Los tipos de crecimiento lento de NHL tienen un pronóstico relativamente bueno, con una mediana de supervivencia de hasta 10 a 20 años, pero habitualmente no son curables en los estadios clínicos avanzados. Los estadios tempranos (estadios I y II) del linfoma no Hodgkin (NHL) de crecimiento lento se pueden tratar eficazmente con radioterapia sola. La mayoría de los tipos poco activos tienen morfología ganglionar (o folicular).
Los tipos de NHL de crecimiento rápido tienen una evolución natural más corta, pero un número significativo de estos pacientes se puede curar con regímenes intensivos de quimioterapia combinada.
El linfoma no Hodgkin (NHL) también puede clasificarse en dependencia de las etapas y categorías
Cuadro 2. Etapas y categorías del linfoma no Hodgkin.
Etapa I: afectación de un grupo de ganglios linfáticos
Etapa II: afectación de dos o más grupos de ganglios linfáticos del mismo lado del diafragma
Etapa III: afectación de grupos de ganglios linfáticos a ambos lados del diafragma
Etapa IV: afectación de uno o más órganos aparte de los ganglios linfáticos y posible afectación de los ganglios linfáticos
A medida que mejoró la comprensión del linfoma no Hodgkin (NHL) y que el diagnóstico histopatológico del LNH se volvió más sofisticado con el uso de técnicas inmunológicas y genéticas, se describió un cierto número de entidades patológicas nuevas. Además, cambió la manera de entender y de tratar muchos de los subtipos patológicos descritos anteriormente. Como resultado, la Working Formulation se tornó obsoleta y menos útil para los médicos y patólogos. Es por eso que patólogos de Europa y los Estados Unidos propusieron una nueva clasificación, la Revised European American Lymphoma (REAL). Desde 1995, los miembros de las sociedades europeas y estadounidenses de hematopatología colaboran en la elaboración de una clasificación nueva de la Organización Mundial de la Salud (OMS), que representa una versión actualizada del sistema REAL.
En la modificación que realizó la OMS de la clasificación REAL, se reconocen tres categorías importantes de neoplasias linfoides malignas con base en la morfología y el linaje celular: neoplasias de células B, neoplasias de células T/citolíticas naturales (CN) y linfoma de Hodgkin. Tanto los linfomas como las leucemias linfoides se incluyen en esta clasificación porque tanto la fase sólida como la fase circulante están presentes en muchas neoplasias linfoides, y la distinción entre ellas es artificial. Por ejemplo, la leucemia linfocítica crónica de células B y el linfoma linfocítico de células B pequeñas son, simplemente, distintas manifestaciones de la misma neoplasia, tal como lo son los linfomas linfoblásticos y las leucemias linfocíticas agudas. Dentro de las categorías de células B y de células T, se reconocen dos subdivisiones: neoplasias precursoras, que corresponden a los estadios tempranos de diferenciación, y las neoplasias diferenciadas más maduras.
Cuadro 3. Clasificación REAL/OMS actualizada
Neoplasias de células B
- Neoplasia de células B precursoras: leucemia linfoblástica aguda de células B precursoras/linfoma linfoblástico (LLB).
- Neoplasias de células B periféricas.
- Leucemia linfocítica crónica de células B/linfoma linfocítico de células pequeñas.
- Leucemia prolinfocítica de células B.
- Linfoma linfoplasmacítico/inmunocitoma.
- Linfoma de células del manto.
- Linfoma folicular.
- Linfoma extraganglionar de células B de la zona marginal de tipo de tejido linfático asociado con mucosa (TLAM).
- Linfoma ganglionar de células B de la zona marginal (± células B monocitoides).
- Linfoma esplénico de la zona marginal (± linfocitos vellosos).
- Leucemia de células pilosas.
- Plasmacitoma/mieloma de células plasmáticas.
- Linfoma difuso de células B grandes.
Neoplasias de células T y de supuestas células CN
Neoplasia de células T precursoras: leucemia linfoblástica aguda de células T precursoras y LLB.
Neoplasias de células T y de células CN periféricas.
Leucemia linfocítica crónica de células T/leucemia prolinfocítica.
Leucemia linfocítica de células T granulares.
Micosis fungoide/síndrome de Sézary.
Linfoma de células T periféricas, sin otra indicación.
Linfoma hepatoesplénico de células T γ/δ.
Linfoma similar a paniculitis subcutáneo de células T.
Linfoma angioinmunoblástico de células T.
Linfoma extraganglionar de células T/CN, tipo nasal.
Linfoma intestinal de células T, tipo enteropático.
Linfoma de células T adultas/leucemia (virus linfotrópico humano de células T [VLHT] 1+).
Linfoma anaplásico de células grandes, tipo sistémico primario.
Linfoma anaplásico de células grandes, tipo cutáneo primario.
- Leucemia de células CN de crecimiento rápido.
Linfoma de Hodgkin
- Linfoma de Hodgkin con predominio linfocítico nodular.
- Linfoma de Hodgkin clásico.
- Linfoma de Hodgkin con esclerosis nodular.
- Linfoma de Hodgkin clásico rico en linfocitos.
- Linfoma de Hodgkin de celularidad mixta.
Linfoma de Hodgkin con agotamiento de linfocitos.
El linfoma linfoblástico es una forma muy dinámica de Linfoma no Hodgkin, que se presenta a menudo en pacientes jóvenes, pero no exclusivamente en estos. El linfoma linfoblástico se relaciona por lo común con masas mediastínicas grandes y tiene una gran predilección por diseminarse hasta la médula ósea y el sistema nervioso central (SNC).
Representa aproximadamente el 2% de todos los casos de linfoma no Hodgkin (NHL). Se presenta más comúnmente en adultos jóvenes y con más frecuencia en hombres que en mujeres. Este tipo de linfoma es igual a una forma de leucemia denominada leucemia linfoblástica aguda (acute lymphoblastic leukemia, ALL). Cuando se encuentra principalmente en los ganglios linfáticos, se denomina linfoma linfoblástico, y cuando afecta principalmente la sangre o la médula ósea, se denomina Leucemia Linfoblástica Aguda (Acute Lymphoblastic Leukemia [ALL]). Cuando afecta principalmente los ganglios linfáticos, los más frecuentemente comprometidos son los del centro del pecho, aunque puede haber toma ganglionar generalizada.
Tanto el linfoma linfoblástico como la ALL son enfermedades agresivas que requieren quimioterapia intensiva, incluido tratamiento para el sistema nervioso central para evitar la diseminación al cerebro.
Los paradigmas de tratamiento se basan en ensayos clínicos para la leucemia linfoblástica aguda (ALL), debido a que el linfoma linfoblástico y la ALL se consideran diferentes manifestaciones de la misma enfermedad biológica.
El tratamiento habitualmente se estructura como el de la Leucemia Linfoblástica Aguda (LLA). La quimioterapia combinada intensiva con profilaxis del sistema nervioso central (SNC) es el tratamiento estándar para este tipo histológico de crecimiento rápido de linfoma no Hodgkin (NHL). A veces,
se administra radioterapia hacia las áreas de masas tumorales voluminosas. Debido a que estas formas de linfoma no Hodgkin (NHL) tienden a evolucionar con rapidez, la quimioterapia combinada se instituye de inmediato una vez que se confirma el diagnóstico.
Las opciones de tratamiento estándar para el linfoma linfoblástico en adultos son las siguientes:
El tratamiento estándar es quimioterapia combinada intensiva con profilaxis del sistema nervioso central (SNC).Radioterapia
A veces, se administra radioterapia dirigida a áreas de masas tumorales voluminosas.
Inmunoterapia
La inmunoterapia (también denominada terapia biológica) ayuda a estimular las defensas naturales del cuerpo para combatir el cáncer. Utiliza materiales producidos por el cuerpo o fabricados en un laboratorio para mejorar, identificar o restaurar la función del sistema inmunológico. Los anticuerpos monoclonales, el interferón y las vacunas son terapias biológicas que se están evaluando en ensayos clínicos para diferentes subtipos de LNH. Cada uno se describe a continuación.
Anticuerpos monoclonales. Los anticuerpos monoclonales están dirigidos hacia una proteína específica y no afectan a las células que no tienen la proteína. El anticuerpo monoclonal llamado rituximab se utiliza para el tratamiento de muchos tipos diferentes de linfoma de células B. El rituximab actúa atacando una molécula llamada CD20 que se encuentra en la superficie de todas las células B y linfomas de células B. Cuando el anticuerpo se une a esta molécula, algunas células del linfoma mueren y otras se vuelven más susceptibles a desaparecer con la quimioterapia. Si bien funciona bien por sí sola, las investigaciones demuestran que funcionó mejor cuando se la combinó con quimioterapia en pacientes con la mayoría de los tipos de linfoma no Hodgkin (NHL) de células B. El rituximab se administra también después de una remisión de linfomas indolentes para aumentar la duración de la remisión.
Otro anticuerpo monoclonal, brentuximab vedotin, se aprobó en el año 2011 para el tratamiento del linfoma anaplástico sistémico de células grandes en pacientes que no se beneficiaron de al menos un tipo de quimioterapia y en pacientes con linfoma de Hodgkin que no se beneficiaron del trasplante de células madre o que no pudieron someterse a un trasplante de células madre. En la actualidad, se está investigando sobre otros anticuerpos monoclonales más nuevos para el linfoma.
Anticuerpos radiomarcados. Los anticuerpos radiomarcados son anticuerpos monoclonales con partículas radioactivas adheridas, cuyo objetivo es concentrar la radiación directamente en las células del linfoma. Estos tipos de medicamentos son nuevos y están en investigación. En general, se cree que los anticuerpos radioactivos son más potentes que los anticuerpos monoclonales regulares, pero más perjudiciales para la médula ósea. Este tipo de tratamiento también se conoce como radioinmunoterapia (RIT).
Trasplante de células madre/médula ósea
Un trasplante de células madre es un procedimiento médico en el cual la médula ósea que contiene cáncer se reemplaza por células altamente especializadas, denominadas células madre hematopoyéticas, que se desarrollan en médula ósea sana. Las células madre hematopoyéticas se encuentran tanto en el torrente sanguíneo como en la médula ósea. Hoy en día, a este procedimiento se lo denomina más comúnmente trasplante de células madre, en lugar de trasplante de médula ósea, porque en realidad lo que se trasplanta son las células madre sanguíneas y no el tejido de la médula ósea
El trasplante de células madre es, con frecuencia, un tratamiento agresivo y generalmente se lo indica únicamente a pacientes con linfoma no Hodgkin (NHL) cuya enfermedad es progresiva o recurrente. Sin embargo, en algunos subtipos de LNH, como el linfoma de células del manto y algunos linfomas de células T, el trasplante de células madre puede recomendarse para evitar la recurrencia.
Existen dos tipos de trasplante de células madre, según el origen de las células madre sanguíneas de reemplazo: alogénico (ALLO) y autólogo (AUTO). Las células que se usan en los trasplantes AUTO provienen de los mismos pacientes, mientras que las que se usan en los trasplantes ALLO provienen de donantes sanos compatibles.
En ambos tipos, el objetivo del trasplante es destruir células cancerosas en la médula, la sangre y otras partes del cuerpo, y permitir que células madre sanguíneas de reemplazo creen una médula ósea sana. En la mayoría de los trasplantes de células madre, el paciente es tratado con dosis altas de quimioterapia y/o radioterapia para destruir la mayor cantidad posible de células cancerosas. Se utilizan dosis altas debido a que los pacientes que se someten a este tratamiento padecen una enfermedad que ha demostrado ser resistente a dosis normales de quimioterapia. Las dosis más altas de quimioterapia son más eficaces contra el linfoma no Hodgkin (NHL) recurrente que las dosis estándar.
Bibliografía.
- Alavanja MCR, Ross MK, Bonner MR. Increased cancer burden among pesticide applicators and other due to pesticide exposure. CA: A Cancer Journal for Clinicians. 2013;63(2):120-142.
- American Cancer Society: Cancer Facts and Figures 2014. Atlanta, Ga: American Cancer Society, 2014.
- Bierman PJ, Harris N, Armitage JO. Non-Hodgkin’s lymphoma. In: Goldman L, Ausiello D, eds. Cecil Medicine. 24th ed. Philadelphia, Pa: Saunders Elsevier; 2011: chap 191.
- Casulo C, Rich L. Linfoma no Hodgkin. Leukemia and Lymphoma Society. Revisado 2014.
- Instituto Nacional del Cáncer. Estados Unidos. Disponible en: http://www.cancer.gov/espanol/pdq/tratamiento/no-hodgkin-adultos/HealthProfessional/page1.
- Kaushansky K, Lichtman MA, Beutler, et al. Williams Hematology. 8va. ed. New York, NY: McGraw-Hill Professional; 2010.
- Leonard JP. Is rituximab maintenance still standard of care in indolent non-Hodgkin lymphoma? Clinical Advances in Hematology & Oncology. 2012;10(8):540-542.
- National Comprehensive Cancer Network. Practice Guidelines in Oncology—v.1.2013. Non-Hodgkin Lympoma. Disponible en: nccn.org/professionals/physician_gls/pdf/nhl.pdf.
- Shankland KR, Armitage JO, Hancock BW: Non-Hodgkin lymphoma. Lancet 380 (9844): 848-57, 2012.