domingo, 24 de agosto de 2025
En un paciente con LLC subyacente, una disminución repentina secundaria a fiebre, linfadenopatía voluminosa y dolor abdominal debe hacer sospechar una transformación de Richter, que es la conversión de leucemia linfocítica de células B bien diferenciada en un linfoma difuso de células grandes. El síndrome de Richter presagia un mal pronóstico.
El diagnóstico de púrpura trombocitopénica idiopática (PTI) en un paciente que presenta trombocitopenia aislada requiere una prueba de VIH, serología de anticuerpos antinucleares y (en pacientes mayores de 60 años) una biopsia de médula ósea para descartar causas secundarias o PTI, que pueden incluir VIH, lupus eritematoso sistémico y síndrome mielodisplásico.
Dos tipos de cáncer que presentan niveles bajos de fosfatasa alcalina leucocitaria (LAP) son la leucemia mieloide crónica (LMC) y la hemoglobinuria paroxística nocturna (HPN). En ocasiones, el síndrome mielodisplásico puede presentarse con un nivel de LAP (Fosfatasa alcalina leucocitaria) de 0 (48-50).
El sangrado que ocurre en pacientes con trastornos plaquetarios difiere del tipo de sangrado encontrado en pacientes con deficiencias de factores. Los primeros tienen sangrado de las mucosas (es decir, sangrado de encías, epistaxis, menorragia y petequias o púrpura). La deficiencia de factores se presenta como sangrado en tejidos más profundos (p. ej., sangrado por hemartrosis en las articulaciones), sangrado en los músculos y sangrado retardado.
Un paciente sin enfermedad hematológica, oncológica (anemia falciforme, LLC) o inmunosupresora subyacente (síndrome de inmunodeficiencia adquirida) que presenta una anemia aislada profunda con una respuesta reticulocitaria prácticamente ausente debe someterse a una tomografía computarizada torácica para descartar la presencia de un timoma. Los timomas con o sin miastenia gravis se asocian con aplasia pura de glóbulos rojos. La clave para el diagnóstico es encontrar una disminución en el recuento de reticulocitos. La confirmación se realiza mediante trepanación de médula ósea que demuestra una disminución o ausencia de progenitores de glóbulos rojos. Otras causas de aplasia pura de glóbulos rojos son idiopática, parvovirus B19, leucemia crónica, VIH y anemia inducida por fármacos.
La enfermedad de Castleman multicéntrica (ECM) es un trastorno linfoproliferativo asociado con la infección por el virus del herpes humano tipo 8 (HHV-8). Las características incluyen fiebre, pérdida de peso, linfadenopatía generalizada y hepatoesplenomegalia. Los pacientes infectados con el virus de la inmunodeficiencia humana (VIH) tienen un mayor riesgo de ECM. En ocasiones, el sarcoma de Kaposi asociado al HHV-8 puede presentarse simultáneamente con ECM.
Fiebre, disnea, infiltrados pulmonares, aumento de peso e hipotensión en un paciente con leucemia promielocítica aguda subyacente que recibe tratamiento con ácido transretinoico deben hacer sospechar el síndrome del ácido retinoico (RAS). Antes de atribuir estos síntomas al RAS, se deben descartar causas infecciosas.
sábado, 23 de agosto de 2025
Claves diagnósticas
Los receptores de trasplantes de órganos sólidos con riesgo de desarrollar trastornos linfoproliferativos postrasplante (PTLD) son aquellos que han sido tratados con ciclosporina y azatioprina, OKT3 o FK506 en dosis altas. Los receptores de trasplantes pulmonares y cardíacos también están en riesgo. Se cree que el virus de Epstein-Barr (VEB) y los receptores que son negativos al VEB y reciben un órgano sólido de un donante positivo al VEB tienen el mayor riesgo de desarrollar PTLD. El primer paso del tratamiento es la reducción de las dosis de los inmunosupresores. El linfoma no Hodgkin de células B afecta característicamente a sitios extraganglionares, con un tropismo único por el órgano trasplantado.
Claves diagnósticas
Si una paciente desarrolla hematomas espontáneos en la piel unos días después de recibir una transfusión de sangre, solicite un recuento de plaquetas para descartar púrpura postransfusional. La trombocitopenia grave en esta afección es causada por anticuerpos dirigidos contra un antígeno específico en las plaquetas, así como plaquetas libres de antígeno.
Claves diagnósticas
El síndrome antifosfolípido (SAF) catastrófico debe considerarse en pacientes con mal aspecto que presentan insuficiencia multiorgánica y trombosis generalizada que a menudo conducen a gangrena. La combinación de insuficiencia renal, síndrome de dificultad respiratoria aguda e infartos cutáneos debe aumentar la posibilidad de un SAF catastrófico.
Claves diagnósticas
Se debe sospechar anemia hemolítica microangiopática en pacientes con hemólisis no inmunitaria (prueba de Coobs negativa) combinada con un frotis de sangre periférica que muestra esquistocitos y células en casco. Los glóbulos rojos están tan fragmentados que el volumen corpuscular medio puede estar reducido. En pacientes no embarazadas, la anemia hemolítica microangiopática (MAHA) y la trombocitopenia deben hacer sospechar PTT o síndrome hemolítico urémico. Un nivel elevado de lactato deshidrogenasa suele estar presente en la MAHA y puede utilizarse, además de los hematocritos seriados, para evaluar la gravedad de la anemia.
Claves diagnósticas
La hipertrofia de las encías puede ser secundaria al uso crónico de fenitoína, ciclosporina o nifedipina, así como a la infiltración leucémica en la leucemia mielomonocítica aguda.
Claves diagnósticas
El diagnóstico del síndrome de anticuerpos antifosfolípidos debe realizarse en pacientes que presentan trombosis arteriales venosas recurrentes, trombocitopenia, pérdida fetal recurrente (clásicamente en el segundo trimestre) o livedo reticularis. Los hallazgos de laboratorio críticos para el diagnóstico deben confirmarse en dos ocasiones con al menos 3 meses de diferencia: anticuerpo anticardiolipolítico Ig M o Ig G, anticoagulante lúpico o anti glucoproteína B2.
Claves diagnósticas
La coagulación intravascular diseminada es una de las consecuencias graves de la leucemia promielocítica aguda (tipo M3 de leucemia mieloide aguda), mientras que la infiltración meníngea es más frecuente en la leucemia monocítica o mielomonocítica aguda (M4 y M5).
Claves diagnósticas
Se debe sospechar tromboangitis obliterante (enfermedad de Buerger) en pacientes menores de 45 años que presentan claudicación en antebrazo, pantorrilla o pie. Los hombres se ven afectados con mayor frecuencia que las mujeres y, aunque se desconoce la causa, el tabaquismo parece ser el factor de riesgo más importante. Dejar de fumar es la modalidad de tratamiento más importante. El examen físico generalmente revela pulsos reducidos o ausentes, a diferencia del síndrome de anticuerpos antifosfolípidos o émbolos de colesterol.
Claves diagnosticas
Un paciente que recibe tratamiento para la púrpura trombocitopénica idiopática (PTI) con IVIG y posteriormente desarrolla insuficiencia renal puede tener nefrotoxicidad asociada a la IVIG
Claves diagnosticas terapeúticas de hematologia
El mieloma múltiple es una de las causas de un anión gap bajo o negativo porque las proteínas monoclonales son catiónicas.
Claves diagnóstico terapeútica de hematologia
El diagnóstico de crioglobulinemia esencial mixta se realiza mediante la historia clínica (síntomas constitucionales, artralgias, púrpura palpable en las extremidades inferiores, fenómeno de Raynaud y hematuria con insuficiencia renal), anomalías de laboratorio (hipocomplementemia a menudo con C4 desproporcionadamente disminuido) y crioglobulinas circulantes. Se observa una fuerte asociación con el virus de la hepatitis C (VHC) y la crioglobulinemia esencial mixta, y la presencia del VHC siempre debe investigarse porque el tratamiento puede dirigirse contra el VHC con antivirales.
Claves diagnóstica terapeútica de hematologia
La pentada de hepatomegalia dolorosa, ictericia, ascitis, hiperbilirrubinemia y aumento de peso dentro de los 21 días posteriores al trasplante de médula ósea debe sugerir el diagnóstico de enfermedad venooclusiva (EVO), una presentación clínica no muy diferente del síndrome de Budd Chiari. El diagnóstico diferencial incluye enfermedad de injerto contra huésped, colestasis de sepsis y malignidad recurrente. La EVO es a menudo un diagnóstico clínico, aunque la biopsia hepática transyugular puede ayudar en el diagnóstico demostrando oclusión venular hepática y necrosis hemorrágica centrolobulillar.
Claves diagnostica terapeútica
Un paciente neutropénico que presenta dolor abdominal en el cuadrante inferior derecho, fiebre y diarrea sanguinolenta debe sospechar tiflitis (colitis necrosante, inflamación del ciego). La tiflitis se presenta con mayor frecuencia en pacientes neutropénicos que han sido tratados por neoplasias hematológicas con agentes citotóxicos. Puede presentarse dolor de rebote en el examen físico y la tomografía computarizada (TC) es un método sensible para demostrar el engrosamiento del ciego. El tratamiento debe comenzar con antibióticos dirigidos contra bacilos gramnegativos y anaerobios (flora intestinal mixta típica). En casos graves, la cirugía es ocasionalmente necesaria.
Claves diagnóstica terapeútica de hematologia
El síndrome POEMS es un acrónimo para una enfermedad en la que los hallazgos predominantes son polineuropatía (sensorial-motora), organomegalia (hepatoesplenomegalia y linfadenopatía), endocrinopatía (amenorrea, disminución de la libido, diabetes mellitus, hipotiroidismo o hipoadrenalismo), pico M y cambios en la piel (hiperpigmentación, hipertricosis e hipocratismo digital). Este síndrome tiene características tanto del mieloma múltiple (pico M y lesiones óseas escleróticas) como de la macroglobulinemia de Waldenström (organomegalia y una polineuropatía desmielinizante), además de las manifestaciones dermatológicas y endocrinológicas.
Claves diagnostica terapeutica de hematologia
En un paciente con angioedema, la presencia de un nivel normal de C4 descarta la posibilidad de edema angioneurótico hereditario (HANE) porque el defecto está en el inhibidor de la esterasa C1 (niveles bajos o disfuncionales) que conduce a la degradación de C4. Si el nivel de C4 es bajo, entonces se deben evaluar los niveles de inhibidor de la esterasa C1 y la actividad funcional además de los niveles de C1q. Los niveles de C1q son normales en el HANE, pero están disminuidos en el linfoma de células B adquirido. La deficiencia del inhibidor de la esterasa C1 se ha asociado con malignidad, en particular, en el linfoma de células B.
Claves diagnostica terapeutica de hematologia
Un paciente hipogammaglobulinémico que desarrolla anafilaxia después de recibir inmunoglobulina intravenosa (IVIG) debe ser investigado para detectar deficiencia de inmunoglobulina A (Ig A). Estos pacientes tienen anticuerpos anti-Ig A que reaccionan con la Ig A en las preparaciones de IVIG que deben usarse con el menor contenido posible de IgA.
lunes, 5 de mayo de 2025
Purpura Senil
Casos Clínicos: Varón de 61años con púrpura en miembro superior.
El Dr. José Cendejas, de Ciudad de México, México, envió estas imágenes con el siguiente texto:


Se trata de un hombre de 61 años, hipertenso controlado en tratamiento con enalapril, hipercolesterolemia en tratamiento con atorvastatina y psoriasis en tratamiento con crema de betametasona (uso crónico). Antecedentes familiares: Hijo con miastenia gravis y púrpura primaria, Padre con púrpura primaria. Acudió por presentar un hematoma relacionado con un golpe mientras jugaba al baloncesto, con una evolución de 9 días sin cambios de color (rojo vinoso) y aumento de tamaño. Análisis de laboratorio: Hb 15.4, Leucocitos 8.34, Plaquetas 247, Reticulocitos 2%, TPT 31.5, TP 10.3, INR 0.84, Dímero D 0.73 ug/ml.
Opinión: Se trata de equimosis en la región dorsal del antebrazo y lamano, con antecedente traumático previo. Si efectivamente como dice la historia, esto tiene 9 días de evolución, no deben plantearse otros diferenciales como angiomas etcétera. Cuando nos encontramos frente a una manifestación de un trastorno hemorrágico, incluyendo los hematomas, equimosis y petequias en la piel, debemos abocarnos a tomar una cuidadosa historia clínica, con registro de antecedentes familiares y personales. En este tipo de situaciones, el interrogatorio del paciente acerca de sus antecedentes desde el nacimiento (sangrado del cordón umbilical), sangrado post parto, sangrado post extracciones o procedimientos dentarios, hemorragias mucosas, sangrado por diversos sistemas (digestivo, genitourinario, respiratorio incluyendo antecedentes de epistaxis, etcétera), suele dar mayor información que cualquier prueba de laboratorio. Por supuesto, el exhaustivo interrogatorio debe ser acompañado de un examen físico completo y un laboratorio, haciendo énfasis en la hemostasia (recuento de plaquetas, investigación de las vías extrínseca e intrínseca de la coagulación, tiempo de sangría), que de acuerdo a los resultados deberán o no ser seguidos por estudios más específicos. En este caso, de la historia se desprende que este paciente tiene ANTECEDENTES DE COAGULOPATÍAS EN SU FAMILIA (padre e hijo con antecedentes de púrpura primaria, que entiendo se refiere a trombocitopenia autoinmune), y USO CRÓNICO DE CORTICOSTEROIDES TÓPICOS (betametasona crema). Habrá que averiguar si existe toma crónica de AINES o aspirina, o anticoagulantes, y una larga lista de fármacos, que también pueden ser causa de equimosis a repetición, espontáneas o como consecuencia de traumas mínimos. En este paciente, el resultado normal de su recuento de plaquetas descarta púrpura trombocitopénica, y la investigación del TP, y el KPTT descartan coagulopatías, déficits de factores de coagulación, etcétera. Hay, sin embargo, cuadros como la enfermedad de Von Willebrand, que puede cursar con pruebas básicas de hemostasia dentro de los límites normales y hay que hacer estudios específicos para demostrarla. Inclusive, existen estudios de funcionalidad plaquetaria (agregación), que pueden poner en evidencia cambios cualitativos de las plaquetas, aun con número de plaquetas normales. A veces algunas deficiencias vitamínicas pueden causar equimosis (por ej déficit de vitamina C). En conclusión, creo que este paciente presentó un hematoma postraumático en piel (equimosis), favorecido por el uso crónico de corticoides, que, aunque sean utilizados en forma tópica, pueden tener efectos sistémicos a través de la absorción a través de la piel, especialmente cuando se usan por períodos de tiempo prolongados. Los corticoides usados crónicamente pueden producir alteraciones en la síntesis de colágeno, así como un grado variable de atrofia dérmica, lo que hace más visibles los vasos de la dermis y el efecto de su sangrado. Por otro lado, hay que decir que a la edad de este paciente, la aparición espontánea de este tipo de lesiones se la conoce como PÚRPURA SENIL, descrita por Bateman, y que justamente se suele ver en esta zona afectada porel fotoenvejecimiento, y atrofia cutánea.
sábado, 5 de abril de 2025
Actualización en mastocitosis
Actualización en mastocitocis
La mastocitosis es la proliferación anormal de mastocitos que puede afectar 1 o varios órganos.
La mastocitosis es la proliferación anormal de mastocitos que puede afectar 1 o varios órganos. Esta célula almacena importantes sustancias farmacológicamente activas. Sus manifestaciones más frecuentes son las cutáneas, pero pueden aparecer otras a causa de afectación de órganos como la médula ósea, hígado, bazo, huesos, ganglios linfáticos y tubo digestivo. Existen varios métodos diagnósticos, pero la histología es fundamental. El tratamiento incluye medidas generales y específicas; actualmente el uso del interferón ha cobrado interés.
En el año 1878 Paul Ehrlich, médico alemán, describió los mastocitos del tejido conectivo y postuló, que estas células podrían estar relacionadas con la inflamación de tejidos, vasos sanguíneos, nervios y focos neoplásicos. Identificó al mastocito como una célula presente en proporción variable en el tejido conectivo y en la mucosa de diferentes órganos: pulmón, tubo digestivo, tejido peridental y subcutáneo, ganglios linfáticos y cápsula del timo.La mastocitosis es una enfermedad caracterizada por la hiperplasia de mastocitos que puede involucrar varios órganos como la piel, tracto gastrointestinal, ganglios linfáticos, médula ósea, hígado, y bazo. El mastocito es una célula hematopoyética derivada de la célula progenitora mieloide pluripotencial. El mastocito o célula cebada almacena importantes sustancias farmacológicamente activas llamadas mediadores, unos se encuentran preformados en los gránulos, mientras otros se sintetizan y se liberan tras un estímulo apropiado
Independiente del órgano involucrado, la patogénesis de la mastocitosis es en gran medida el resultado de la activación de mastocitos, llevando a la secreción aumentada de los mediadores celulares de los mastocitos. Hay tres categorías de mediadores celulares de los mastocitos: 1) citoquinas proinflamatorias ([alpha]) y [beta], interferon, factor de crecimiento) mediadores de degranulación asociados (histamina, heparina, proteasas), y 3) derivados lipídicos ( leukotrienes, LTC4, LTE4, prostaglandina D2, factor activador de plaquetas). Los síntomas y signos de la enfermedad pueden estar en relación con la liberación de mediadores mastocitarios o con la infiltración orgánica por estas células. Existe una gran heterogeneidad en cuanto a las manifestaciones clínicas, incluso dentro de una misma forma de la enfermedad, que puede ir desde la forma indolente hasta la sistémica, que es la más grave. Los efectos de estos mediadores incluyen prurito, urticaria vasodilatación, vasopermeabilidad aumentada, hipersecreción gástrica, broncoespasmo, anticoagulación local, osteoporosis, y lesiones óseas.
La mastocitosis puede ocurrir a cualquier edad y puede tener un ligero predominio en el varón a (1.5:1.0). Su predominio es desconocido, y las forma familiar es rara.
La mastocitosis cutánea presenta cuatro modelos:
1. Urticaria pigmentosa que es muy común en la niñez apareciendo como máculas rosa-castaño en el tronco, normalmente tiene un curso favorable y un compromiso sistémico menor.
2. Mastocitomas
3. Mastocitosis cutánea difusa que es muy raro y aparece en la infancia, dando un color anaranjado a la piel; el compromiso sistémico es más común
4. Telangiectasia macularis que normalmente aparece en los adultos.
En el adolescente y en el adulto las dos manifestaciones clínicas típicas de la mastocitosis son el prurito cutáneo y el flush, este último se caracteriza por enrojecimiento facial y de la zona superior del tronco con sensación de calor sin sudación y puede ir acompañado por palpitaciones, dificultad respiratoria, dolor torácico, cefalea, en ocasiones pérdida de la conciencia de breve duración, hipotensión, hipertensión, taquicardia. Este cuadro puede aparecer de forma espontánea o desencadenado por calor, estrés, ejercicio físico, menstruación y ciertos medicamentos como la aspirina, los antiinflamatorios no esteroideos y opiáceos. Agentes físicos (calor y frío).
Picaduras de abejas y avispas. Alimentos (quesos, vinos, mariscos, chocolate, tomate, platános). Estudios radiológicos de contrastes.
En estos pacientes predominan los cuadros de dolor abdominal acompañados de diarrea, náuseas, vómitos, a veces fiebre y astenia.
No es infrecuente que las formas indolentes estén libres de síntomas o sólo presenten prurito ocasional y cuadros de flush, generalmente desencadenados por diversos agentes. En las formas agresivas puede aparecer cansancio, disnea, sudación nocturna, pérdida de peso, o incluso síntomas neuropsiquiátricos como convulsiones, alteraciones de la conciencia o síntomas depresivos. En el niño pequeño la evolución se efectúa en 2 fases, un brote de pápulas urticarianas pruriginosas de color rosado o rojo, que evolucionan al carmelita y cuyo número va aumentando progresivamente en algunos meses. Después las lesiones se van estabilizando bajo la forma de manchas pigmentadas permanentes, ligeramente abultadas o no, y pueden ocurrir brotes congestivos, pero sin la aparición de nuevos elementos; en el niño mayor también pueden estar presentes los síntomas descritos en el adulto.
Los mastocitomas son lesiones solitarias que miden 1 a 5 centímetro de diámetro que puede aparecen en cualquier sitio. El ataque generalmente se inicia antes de los 6 meses de edad, pero las lesiones pueden estar presentes en el nacimiento. Inicialmente, los mastocitomas se presentan como una bulla recurrente o pápula evanescente. Después, puede desarrollar una placa infiltrado rosa o amarillo, o de color canela. La superficie adquiere una textura de cáscara de naranja y la hiperpigmentación pueden ser prominente. En la mayoría de los casos, hay involución espontánea. El raspado de las lesiones o el trauma del mastocitoma puede producir la descarga de histamina, llevando a la urticaria (Darier) o a veces a la formación de la ampolla. Los pacientes con cualquiera de los cuatro tipos de mastocitosis pueden experimentar colapso vascular. De vez en cuando, tales reacciones pueden ser provocadas por el alcohol, aspirina, los narcóticos, medios de contraste iodados, picaduras de insectos, ejercicio, o infección. El mastocitoma cutáneo raramente se acompaña colapso. Afortunadamente, la mayoría de los pacientes con mastocitoma tienen lesiones superficiales que son reconocibles en el examen físico. El diagnóstico se establece entonces por la biopsia superficial. En los pacientes sin lesiones superficiales que se sospecha mastocitosis, elevados niveles de histamina en plasma u orina y metabolitos de los otros mediadores pueden ser útil para el diagnóstico.
El objetivo primario de tratamiento es controlar lignos y síntomas inducidos por la activación del mastocito. Deben administrarse antihistamínicos locales y sistémicos. Pueden ser beneficiosos los antagonistas del receptor H1 y H2. El cromoglicato disódico puede tener algo de eficacia pero sólo cuando hay compromiso gastrointestinal. Deben aconsejarse a los padres evitar la fricción e irritación de mastocitoma y abstenerse de dar al niño alimentos (quesos, vinos, mariscos, chocolate, tomate, platános) capaces de provocar la degranulación del mastocito. Si a pesar de las medidas anteriores persisten los síntomas debe iniciarse tratamiento con aspirina, y comenzar con dosis bajas que se incrementarán hasta alcanzar dosis necesarias para frenar la síntesis de prostaglandinas, por los mastocitos que suelen alcanzar los 6 g diarios; este tratamiento debe ser hospitalizado bajo estricta vigilancia. Una alternativa a la aspirina es el empleo de antiinflamatorios no esteroideos o el ketotifeno. Existen formas agresivas en donde puede ser necesaria la esplenectomía, que está indicada en los casos que evolucionen con esplenomegalia, especialmente si se acompañan de citopenias. El interferón alfa es capaz de disminuir la infiltración mastocitaria y frenar la liberación de mediadores, al menos en un cierto número de casos. Debe tenerse en cuenta que la terapia con interferón puede desencadenar shock anafiláctico y también existe la posibilidad de desarrollar anticuerpos antiinterferón a largo plazo. Inmunosupresores como la ciclosporina junto con el interferón.Las formas pediátricas son autolimitadas y regresan espontáneamente en la mayoría de los casos. En el adulto las formas indolentes y cutáneas localizadas tienen mejor pronóstico. La edad superior a 50 años es considerada un factor de mal pronóstico. El sexo femenino tiene peor pronóstico, así como las mastocitosis agresivas.
* El Dr. Edgardo Checcacci es editor responsable de IntraMed en la especialidad de Pediatría.
miércoles, 12 de marzo de 2025
Mastocitosis sistemica
An. Med. Interna (Madrid) vol.25 no.3 mar. 2008
Introducción
La mastocitosis sistémica (MS) es una enfermedad clonal de los progenitores mastocíticos de la médula ósea. El cuadro clínico en la MS varía desde una forma asintomática (indolente) a una forma altamente agresiva con una supervivencia muy corta (leucemia de mastocitos) (1).
En 1869, Nettleship describió la urticaria pigmentosa (UP) y en 1887, Unna describió un amento en el número de mastocitos en la UP. Fue ya en 1949, cuando Ellis describió una enfermedad sistémica asociada a hiperplasia de mastocitos.
La mayoría de los pacientes afectos son adultos, aunque la enfermedad puede ocurrir a cualquier edad. En función del momento de aparición de los síntomas, en especial, de las lesiones cutáneas, se define una mastocitosis de la infancia (antes de la pubertad) y una mastocitosis de la edad adulta (después de la pubertad). En un reducido número de casos, se puede detectar una historia familiar (mastocitosis familiar).
Los síntomas derivan de los productos secretados por los mastocitos o de la infiltración de órganos por los mismos. Datos recientes sugieren que la MS se comporta como una enfermedad mieloproliferativa que involucra a los progenitores hematopoyéticos pluripotenciales. Esta idea está potenciada por la existencia de mutaciones en c-kit-816 (2).
Es de especial importancia que se sospeche una mastocitosis sistémica en pacientes con inestabilidad vascular inexplicable, un shock anafiláctico sin causa conocida, enrojecimiento cutáneo y facial idiopático, diarrea, cefaleas y otros síntomas que podrían estar relacionados con la secreción de mediadores (3).
Principios básicos
Los mastocitos derivan de sus progenitores hematopoyéticos y, tanto en humanos, como en roedores, su principal factor de crecimiento es el factor de células stem (SCF) (4).
Los mastocitos que están destinados a residir en tejidos periféricos, como la piel, pulmón e intestino, se originan en la médula ósea y circulan en la sangre como células precursoras CD34+ que posteriormente atraviesan las células endoteliales para depositarse en los tejidos periféricos, donde se diferencian y maduran (5). Allí, forman gránulos de secreción, y expresan en su superficie otros receptores (FcεRI).
Son múltiples las citoquinas y los factores que contribuyen a la mastopoyesis. Una citoquina fundamental en este proceso es el factor de las células stem (CSF). Éste induce el desarrollo de los mastocitos a partir de sus progenitores y promueve su diferenciación terminal y su maduración (6). Los efectos de SCF sobre los mastocitos y sus progenitores está mediado a través de KIT, un receptor transmembrana del tipo de tirosin-kinasa codificado por el proto-oncogen c-kit (7). Los ratones de laboratorio que carecen de c-kit o del gen de SCF, carecen de mastocitos. Por todo esto, se sabe que SCF y KIT son unas moléculas críticas en la regulación del desarrollo de los mastocitos, su proliferación y su supervivencia. Por este motivo, las mutaciones que impliquen una ganancia de función en el gen c-kit se asocian con un mayor crecimiento de los mastocitos. Estas mutaciones, en especial, la c-kit D816V (Asp-816-Val), se identifican con frecuencia en los pacientes con mastocitosis sistémica (8-10).
Al contrario de lo que ocurre con los mastocitos normales, los mastocitos de las mastocitosis sistémicas normalmente expresan CD25 y CD2; las células más inmaduras también podrían expresar otros antígenos adicionales. Los progenitores más inmaduros (tanto los normales como los neoplásicos), expresan CD34 y CD13, así como CD117 (11).
Clasificación
Basándonos en los hallazgos clínicos y en los síntomas, se puede describir cuatro grandes grupos de pacientes con mastocitosis sistémica (Tabla I).
En la mayoría de los pacientes (80%), la mastocitosis sólo afecta a la piel. En el resto de los casos, puede estar infiltrada la médula ósea, el bazo, los ganglios o el tracto gastrointestinal.
En la mastocitosis de la médula ósea, un tipo especial de mastocitosis, la enfermedad está restringida a dicho órgano; es una forma indolente de mastocitosis sin lesiones cutáneas. En estos pacientes, los niveles de triptasa sérica son normales o muy bajos, en contraste a las mastocitosis malignas (12).
Fisiopatología y clínica
Los mastocitos y los basófilos tienen algunas características que los hacen similares, pero también presentan numerosas diferencias. Mientras que el CSF es el principal factor de crecimiento para los mastocitos, la interleukina 3 (IL-3) lo es para los basófilos humanos, que completan su maduración en la médula ósea y entran al torrente sanguíneo cuando ya tienen gránulos de secreción en su interior, receptores de superficie tipo FcεRI y una pequeña cantidad de receptores Kit. Estos circularán por sangre, y solo se depositarán a los tejidos que sufran un proceso inflamatorio (13).
En humanos, los gránulos de secreción de los mastocitos contienen altas cantidades de triptasa, histamina y heparina, quimasa y proteoglicanos de condroitín sulfato E. Una subpoblación de mastocitos también contiene cantidades altas de catepsina G y carboxipeptidasa. Aquellos mastocitos que sólo tienen triptasa se denominan células MCT.
Los basófilos contienen cantidades de histamina similares a la de los mastocitos. Sin embargo, a diferencia de estos, los basófilos contienen sulfato de condroitina tipo A en sus gránulos, así como proteínas eosinofílicas, pequeñas cantidades de triptasa y una proteína aún no bien caracterizada (14,15).
Estos criterios de distinción entre mastocitos y basófilos aún no están demasiado claros. Se ha aislado células metacromáticas (presumiblemente basófilos) de sangre periférica de pacientes con asma activa y con reacciones de hipersensibilidad que eran fuertemente positivas para triptasa, quimasa y Kit de superficie, lo que sugiere que los basófilos de la sangre periférica podrían adquirir patrones de mastocitos bajo determinadas situaciones clínicas (16).
La clínica de la mastocitosis sistémica (MS) se debe a que los tejidos están ocupados por la masa de mastocitos y estos reaccionan mediante fibrosis y liberación de sustancias activas que actúan a nivel local (urticaria pigmentosa, dolor abdominal cólico, gastritis, úlcera péptica) y a nivel sistémico (cefalea, prurito, colapso vascular o rubefacción). Estas manifestaciones clínicas pueden agravarse por la toma de AINEs, alcohol o narcóticos del tipo de la codeína, o durante una anestesia general (Fig. 1).
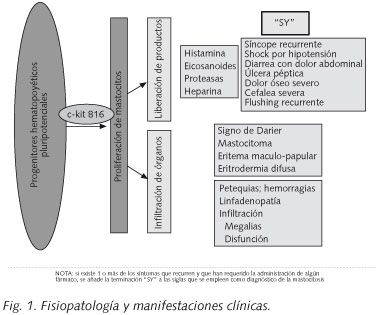
La urticaria pigmentosa es la manifestación cutánea más típica y más frecuente de la mastocitosis, tanto en niños como en adultos. Las lesiones son máculas de pequeño tamaño de color amarillento-rojizo. En casos más raros, pueden ser nodulares. La segunda forma más frecuente de mastocitosis cutánea es la mastocitosis cutánea difusa. Afecta a todo el espesor de la piel, lo que produce un engrosamiento de la misma. Los más jóvenes con urticaria pigmentosa o con mastocitosis cutánea difusa pueden presentar erupciones y bullas con hemorragias asociadas (17).
El examen macroscópico de la piel puede mostrar lesiones típicas que en ocasiones son casi diagnósticas: signo de Darier, mastocitoma, eritema maculo-papular, eritrodermia difusa. Sin embargo, en cada caso, el diagnóstico se puede confirmar mediante una biopsia. La ausencia de lesiones cutáneas no confirma el diagnóstico de mastocitosis. En la enfermedad avanzada, puede detectarse una tendencia al sangrado (petequias espontáneas y/o hematomas), y son el resultado de la trombocitopenia y/o una alteración de la coagulación (por consumo o por hiperfibrinolisis).
El examen físico revela organomegalia en algunos pacientes con MS: linfadenopatía palpable, hepatomegalia y/o esplenomegalia. La infiltración de los órganos por los mastocitos puede conducir, no sólo a organomegalia, sino también a una función anómala de dicho órgano y el diagnóstico se puede confirmar mediante una biopsia.
Los hallazgos clínicos típicos (hallazgos-C) son: la malabsorción con pérdida de peso, hepatomegalia con ascitis, esplenomegalia con hiperesplenismo o fracturas patológicas secundarias a osteolisis (espontáneas). En la enfermedad severa (de alto grado), puede ocurrir anemia, trombocitopenia e infecciones recurrentes secundarias a insuficiencia medular (Tabla II). La ascitis es más frecuente en los casos de mastocitosis asociada a otra anomalía hematológica, y es de mal pronóstico y de difícil control. También puede producirse hipertensión portal, que se puede manejar mediante un shunt porta-cava. Cuando el hiperesplenismo conduce a una intensa pancitopenia, se recomienda hacer una esplenectomía.

La afectación ósea en la mastocitosis está relacionada con la infiltración de la médula ósea por los mastocitos, y está presente en el 70% de los casos. Radiológicamente, puede existir tanto lesiones líticas como blásticas, y, en ocasiones, coexisten ambas. La liberación de histamina por los mastocitos induce esclerosis, lo que explica la presencia de lesiones osteoblásticas, y la liberación de heparina y de prostaglandicnas induce lisis. Esto obliga a hacer el diagnóstico diferencial con otros procesos tales como la mielofibrosis, la enfermedad de Paget, las metástasis óseas o la fluorosis, en el caso de lesiones blásticas, o la osteoporosis quística, la talasemia o la enfermedad de Gaucher en las lesiones líticas (18). Las lesiones son difusas normalmente (85%), aunque también pueden ser focales (5%) o mixtas (10%). En la mayoría de los casos, las lesiones líticas son de pequeño tamaño, inferiores a 0,5 cm y no se acompañan de síntomas. Las lesiones difusas predominan en el esqueleto axial y típicamente combinan áreas de esclerosis con otras líticas (19). En unos pocos casos, sin embargo, se produce fracturas patológicas espontáneas secundarias a osteolisis extensa (20).
Estadificación complementaria
Dependiendo de la edad del paciente, los hallazgos clínicos y los parámetros de laboratorio, la estadificación de la mastocitosis debería incluir las siguientes pruebas: radiografía de tórax, serie ósea, ecografía abdominal y endoscopia digestiva, con toma de biopsia. La ecografía abdominal o la tomografía axial computerizada pueden revelar hepatomegalia, esplenomegalia o linfadenopatías, y sólo se solicitan en los casos en los que se sospeche una mastocitosis agresiva. La serie ósea puede mostrar la presencia de signos de osteoporosis, osteosclerosis o áreas de osteolisis focal. En la mayoría de los casos, las lesiones osteolíticas son pequeñas, y no se acompañan de síntomas clínicos. En otros casos, se puede producir fracturas patológicas espontáneas. Recientemente, Brumsen et al han publicado un artículo en que concluyen que el 9% de los casos de osteoporosis en hombres son secundarios a mastocitosis. El diagnóstico definitivo de muchas de las "osteoporosis idiopáticas" en varones podría, por tanto, conseguirse mediante un estudio anatomo-patológico de la médula ósea o incluso midiendo los niveles del metabolito N-metilhistamina en orina de 24 horas (21, 22).
Histología e inmunohistoquímitca
Para el estudio histológico de tejidos en los que se sospeche infiltración por mastocitos, se requiere de tinciones apropiadas, como la tinción de Giemsa, la tinción con azul de toluidina y tinciones con anti-triptasa. Junto a esto, los mastocitos pueden hacerse visibles empleando la reacción de la cloroacetato esterasa (CAE), enzima que no es exclusiva de los mastocitos (también puede detectarse en los neutrófilos) (23).
El diagnóstico de mastocitosis se basa tradicionalmente en la demostración de acúmulos focales de mastocitos con histología típica y determinadas propiedades citomorfológicas. Sin embargo, a veces resulta difícil diagnosticar la mastocitosis mediante la histología rutinaria.
La médula ósea es el órgano sistémico que se afecta con más frecuencia; el examen histológico de ésta puede demostrar datos típicos (Tabla III). Se recomienda el uso de la tinción de Giemsa y del azul de toluidina en los aspirados de médula ósea de los pacientes sospechosos. En algunos pacientes, el porcentaje de mastocitos en el aspirado de médula ósea es inferior al 5% e incluso, en ocasiones, inferior al 1%. Un porcentaje superior al 20% es casi diagnóstico de la leucemia de mastocitos.
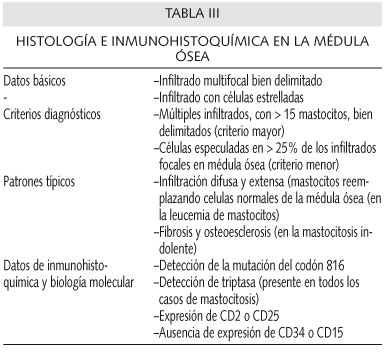
Al contrario de lo que ocurre en la médula ósea, estos criterios diagnósticos no se pueden aplicar a otros órganos. La presencia de algunos mastocitos en el bazo pueden ser suficientes para que se sospeche una mastocitosis, ya que normalmente no suele haber mastocitos en este órgano. Sin embargo, en otros órganos, como el tracto G-I, en los que la presencia de mastocitos es más frecuente, la detección de infiltrados densos de mastocitos no conduce directamente al diagnóstico de mastocitosis sistémica. Con indiferencia del órgano afecto, un infiltrado multifocal bien delimitado, o un infiltrado de células estrelladas, es diagnóstico de MS.
La triptasa y otros parámetros de laboratorio
Un marcador bien establecido y muy importante para el diagnóstico de mastocitosis sistémica son los niveles de triptasa. De hecho, los niveles séricos totales de triptasa reflejan la masa tumoral de mastocitos. En los pacientes sin afectación sistémica, los niveles son normales o están sólo levemente elevados; niveles elevados de triptasa aumentan la posibilidad de que se trate de una mastocitosis sistémica con afectación multiorgánica (la mayoría de los pacientes con mastocitosis sistémica tienen niveles de triptasa superiores a 20 ng/ml). En neoplasias mieloides, en especial, la leucemia mieloide crónica, también se han detectado niveles elevados de triptasa, e incluso pueden elevarse durante una reacción alérgica, por lo que no se trata de un marcador totalmente específico. Basándose en estas limitaciones, Valent et al (20), proponen como criterio diagnóstico de mastocitosis sistémica unos niveles de triptasa elevados de forma persistente, y Schwartz propone que para tener una alta sospecha de mastocitosis, no basta con que la triptasa sérica sea ≥ 20 ng/ml, sino que la relación entre ésta y la β-triptasa también sea ≥ 20, en ausencia de otros trastornos mieloides. En las anafilaxis, están elevadas tanto la triptasa sérica total (α y β), como la β-triptasa, pero la relación entre la total y la segunda es ≤ 10 (24). Aún se necesita más estudios para discernir si este criterio es más sensible o no que el hallazgo de mastocitos en médula ósea.
Aunque el nivel absoluto de triptasa sérica no predice la severidad de la enfermedad, puede proporcionar un método práctico para hacer un seguimiento de la masa de mastocitos tumorales a lo largo del tratamiento de la enfermedad. Además de la triptasa sérica, otros parámetros que pueden emplearse se basan en el hemograma, la bioquímica y la coagulación. En la enfermedad de bajo grado sin AHNMD, estas pruebas debieran ser totalmente normales, mientras que en la enfermedad agresiva o de alto grado, o en la AHNMD, pueden detectarse algunas anomalías, como citopenia, leucocitosis, eosinofilia, basofilia, monocitosis, LDH elevada, elevación de las enzimas hepáticas, hipoalbuminemia, alteración de los parámetros de la coagulación e incluso, mastocitos circulantes.
Mutaciones somáticas de C-Kit
Estudios con cultivos in vitro han demostrado que los mastocitos humanos derivan de células stem pluripotenciales CD34+ y que el factor de las células madre (SCF) es el factor de crecimiento fundamental en la proliferación, diferenciación y supervivencia de los mastocitos.
En la mayoría de los pacientes con MS de inicio en edad adulta se detecta una mutación de c-kit (mutación en el codón 816: Asp-816 → Val: D816V) (25). Por el contrario, la mastocitosis de la infancia no suele asociarse a dicha mutación, aunque se ha detectado en algunos casos pediátricos aislados, al igual que la mutación c-kit Gly-839 → Lys. Esto indica que la MS del adulto y la pediátrica son dos entidades totalmente distintas (26).
Curiosamente, en los pacientes con MS, la mutación c-kit no sólo se detecta en los mastocitos, sino que también puede detectarse en otras líneas celulares, incluyendo los monocitos o incluso las células B. En los casos con mieloproliferación asociada (estado asintomático), la mutación está presente incluso en las células mononucleares de la sangre periférica. En un subgrupo de pacientes con una variante "smoldering" de la mastocitosis, la mutación no sólo se detecta en los mastocitos, sin también en las células que no pertenecen a la línea de las células hematopoéticas de los mastocitos (10).
El análisis de la heterogeneidad de esta enfermedad y ejemplos específicos de cómo progresan, sugieren que debe haber al menos 4 posibilidades que expliquen la diversidad de las manifestaciones clínicas en pacientes con mastocitosis. La primera de estas posibilidades es que una mutación activadora en el codón 816 sea efectiva para promover la división exagerada de mastocitos en algunos individuos, en parte, debido al polimorfismo genético del huésped. Otra posibilidad es que una mutación activante en la posición 816 actúe como una "mutación permisiva" que permita que la enfermedad se desarrolle si tiene lugar una segunda mutación (o incluso una tercera) en vías críticas. En algunos pacientes, puede haber una segunda mutación independiente de la primavera en otro grupo de células, originando un segundo clon maligno. La última posibilidad analizada es que exista inestabilidad cromosómica que predisponga al paciente tanto a la mutación 816 como a otros eventos (27).
Diagnóstico y criterios definitorios
Se ha formulado una serie de criterios mayores y menores, basados en hallazgos clínicos, histológicos, inmunohistoquímicos y de laboratorio, con el fin de diagnosticar una mastocitosis sistémica y de discriminar entre este diagnóstico y el de mastocitosis cutánea, mastocitoma solitario, sarcoma de mastocitos, hiperplasia de mastocitos sistémica y aumento de mastocitos en una neoplasia mielomatosa sin mastocitosis (Fig. 2). El diagnóstico de mastocitosis sistémica se consigue cuando se cumple un criterio mayor y uno menor, o si coexisten los 3 criterios menores. Una vez llegado al diagnóstico, el porcentaje de infiltración de mastocitos en la médula ósea permitirá saber si se trata o no de una leucemia de mastocitos (> = 20% de infiltración). Cuando la infiltración sea inferior al 20%, si no existe hallazgos B ni C, se hablará de mastocitosis indolente; si sólo hay hallazgos B, mastocitosis sistémica, y cuando sólo haya hallazgos C, mastocitosis sistémica agresiva.

En los niños con mastocitosis, el principal hallazgo es la lesión cutánea, que suele desaparecer a lo largo de la adolescencia; por este motivo, si el niño no tiene organomegalia ni una alteración importante en sangre periférica, no será necesario tomar una biopsia de médula ósea. (20). Sin embargo en adultos, incluidos aquellos en los que la mastocitosis se haya iniciado antes de la pubertad, el estudio diagnóstico debe incluir una biopsia de médula ósea (28).
Tratamiento de la mastocitosis sistémica
En la actualidad, no existe tratamiento curativo de la mastocitosis. Los pacientes con mastocitosis indolente suelen tener una progresión lenta a lo largo del tiempo. Según las actuales recomendaciones, estos pacientes deben ser monitorizados de forma estrecha y cuidadosa, pero no se deben tratar con citostáticos.
En los pacientes con mastocitosis sistémica y lesiones óseas significativas, una opción razonable consiste en el tratamiento combinado de IFN-alfa-2b (subcutáneo) y prednisolona (oral) (29). Normalmente se inicia el tratamiento con prednisolona (50-75 mg por día) durante algunos días, y después, se añade IFN-alfa2b adicional comenzando con una dosis de 3 millones de unidades internacionales (UI) tres veces a la semana. Durante los primeros días de tratamiento con IFN, los pacientes deben ser monitorizados en el hospital. Después de unas pocas semanas, la dosis de IFN puede aumentarse 3-5 millones de unidades por día y la prednisolona, reducirse hasta alcanzar una dosis de mantenimiento de 12,5 mg/día o inferior, incluso hasta suspenderse. En los pacientes con una osteoporosis severa y fracturas patológicas, parece preferible la administración de IFN-alfa-2b sin glucocorticoides, dados los efectos secundarios de los mismos. En los pacientes que respondan, el tratamiento debería mantenerse hasta que haya signos o síntomas de toxicidad o efectos secundarios o que exista evidencia de progresión. En una revisión de la literatura disponible, Valent y cols. (30) describieron 14 pacientes con mastocitosis agresiva tratados con IFN-alfa con o sin prednisolona. De estos, el 21,4% (3 pacientes) presentaron una respuesta mayor con desaparición de los hallazgos-C. En cinco pacientes (35,7%) se obtuvo una respuesta parcial, y el 42,9% de los pacientes no respondió. Las tasas de respuestas fueron similares en los que recibieron la combinación con corticoides que en los que no los recibieron. Se ha descrito algún caso de respuestas casi completas en pacientes tratados con IFN-alfa a altas dosis durante largos periodos de tiempo, con una tolerancia aceptable al mismo (31).
Cuando el IFN no es suficiente para el control de la enfermedad, se emplea una amplia variedad de citostáticos (cladribina, arabinósido de citosina, daunorrubicina o hidroxiurea) (32).
Si la situación lo permite y se produce una respuesta mayor a la quimioterapia de inducción, estos pacientes son subsidiarios de un transplante de médula ósea. Si no se puede aplicar el transplante de médula ósea, los pacientes en remisión deberían recibir tratamiento de consolidación.
Durante el tratamiento con quimioterapia, la monitorización se hará basándose en una serie de parámetros: los relacionados con los hallazgos-C (elevación de enzimas hepáticas, la elevación del calcio en sangre o el descenso de la hemoglobina) y los que reflejan la carga tumoral de los mastocitos neoplásicos (la histología de la médula ósea, los niveles de triptasa, CD25). En función a estos datos, se habla de respuesta clínica pura cuando sólo se produzca mejoría en los parámetros relacionados con los hallazgos C; de respuesta completa, cuando se normalicen ambos grupos de parámetros; y de ausencia de respuesta cuando ocurra lo contrario (30).
En la actualidad se está empleando algunos agentes derivados de la biología molecular. Así, por ejemplo, se ha empleado fármacos tales como el Imatinib (STI571), que ha demostrado ser útil en los casos en los que exista determinadas mutaciones, como la c-kit Phe522Cys, pero no útil en otras como la Asp816Val, que es la más frecuente en estos tumores (33).
Fármacos complementarios al tratamiento quimioterápico
A todos los pacientes con mastocitosis agresiva se les administra antagonistas anti-H1 y anti-H2, y/o prednisolona previo al inicio del tratamiento con los agentes quimioterápicos, ya que los mediadores de los mastocitos pueden causar síntomas significativos (34). Estos pacientes se monitorizan cuidadosamente en el hospital durante los primeros días de tratamiento. En los pacientes con síntomas severos y recurrentes asociados a los mediadores, con riesgo de desarrollo de un shock cardiovascular, se recomienda que se disponga de epinefrina para su autoadministración por si fuera necesario.
El tratamiento de la enfermedad intestinal es variable. Los anticolinérgicos pueden proporcionar cierto alivio. La cromolina sódica oral puede ser útil en el manejo de los síntomas abdominales. Los glucocorticoides sistémicos son efectivos en pacientes con malabsorción severa. También la ascitis exudativa ha sido manejada con éxito mediante glucocorticoides sistémicos.
Leucemia de mastocitos
La leucemia de mastocitos es una rara enfermedad caracterizada por una rápida expansión de mastocitos en la médula ósea, la sangre y en otros órganos, con la consiguiente organopatía (35). El pronóstico es grave, y la supervivencia, mínima. No hay ningún tratamiento efectivo para estos pacientes. Incluso la quimioterapia convencional no produce una respuesta duradera en estos pacientes, por lo que se está empleando regímenes agresivos, como los utilizados en la leucemia mieloide aguda. Si existe un donante disponible y se ha obtenido una remisión después de la quimioterapia, también son candidatos a transplante de médula ósea.
Neoplasias malignas hematológicas asociadas
Aproximadamente en un 15-30% de los pacientes con MS se observa una neoplasia hematológica asociada, como síndromes mieloproliferativos, síndromes mielodisplásicos o leucemia mieloide aguda. A veces resulta difícil distinguir entre la neoplasia hematológica asociada y la mastocitosis. La opción terapéutica más recomendada consiste en planificar el tratamiento de la neoplasia maligna hematológica como si no existiera MS asociada, y el tratamiento de la MS como si no existiera la otra patología, y posteriormente, adaptar el tratamiento; en ocasiones, el tratamiento para ambos tratamientos resulta ser el mismo.
Conclusiones
La mastocitosis sistémica es un proceso más frecuente de lo que realmente detectamos en la clínica. Incluso en pacientes con una mastocitosis típica, la mediana de tiempo desde el inicio de los síntomas hasta el diagnóstico es de 1 año. Se debe sospechar ante cualquier paciente con síncopes repetitivos, en especial si no existe otra causa que lo justifique, en varones con osteoporosis y en pacientes con lesiones óseas osteblásticas y/u osteolíticas de etiología no filiada. En estos casos, el primer examen a realizar es la determinación de niveles sanguíneos de triptasa, que, por sí solos, no serían diagnósticos de esta enfermedad. Si dichos niveles permanecieran elevados pasadas algunas semanas, posteriormente es mandatorio hacer un estudio de médula ósea. En ocasiones, un diagnóstico precoz puede evitar situaciones que desencadenen posteriormente un shock a los pacientes afectos. El estudio de médula ósea, junto con una alta sospecha clínica, son la base para el diagnóstico precoz y definitivo.
Eosinofilia
Autor/a: Fei Li Kuang
Fuente: Med Clin N Am 104 (2020) 114.

Introducción |
Los médicos pueden hallar eosinofilia en la sangre o los tejidos, en un hemograma completo que tenga un recuento diferencial de los leucocitos o en una patología tisular.
En este artículo se hace una revisión de la biología de los eosinófilos y las definiciones de eosinofilia en sangre. También se discuten las dificultades para definir la eosinofilia tisular como así las condiciones asociadas con eosinofilia y se analiza un enfoque general para su evaluación.
Los desafíos futuros incluyen determinar qué enfermedades asociadas a los eosinófilos podrían beneficiarse de la terapia dirigida a los eosinófilos, y la identificación de biomarcadores para la actividad de la enfermedad y el diagnóstico.
| Biología de los eosinófilos |
Los eosinófilos son células mieloides, es decir, son células que surgen de la médula ósea y se liberan ya maduros, circulan en la periferia y se convierten en células residentes en los tejidos. El desarrollo de los eosinófilos depende de varias citocinas, incluida las interleucina (IL) IL-5, IL-3 y el factor estimulante de las colonias de granulocitos y macrófagos (GM-CSF, por sus siglas en inglés).
La IL-5 también es fundamental para la activación y supervivencia de los eosinófilos. Aunque éstos se encuentran en la circulación, se cree que son principalmente células que habitan en los tejidos, donde son 100 veces más abundantes. En la sangre, los eosinófilos tienen una vida media de 8-18 horas después de salir de la médula ósea. No está claro cuánto tiempo viven en diferentes tejidos y es posible que su supervivencia pueda extenderse/mantenerse debido a factores exógenos como la IL-5.
| Gránulos de los eosinófilos |
Las micrografías electrónicas revelan que los eosinófilos contienen múltiples tipos de gránulos: primarios, secundarios (específicos de los eosinófilos), pequeños y microgránulos (vesículas secretoras).
Los gránulos primarios son redondos y uniformemente densos, y están compuestos de proteína de cristal de Charcot-Leyden (galectina-10), que forma los bien conocidos cristales de Charcot-Leyden asociados con el esputo de pacientes con asma, descrita años antes del descubrimiento de los eosinófilos. A pesar de esta conocida asociación, ahora se aprecia que estos cristales no son patognomónicos del asma, y que pueden formarse en cualquier lugar en el que haya un exceso de recambio de eosinófilos.
Los gránulos secundarios o específicos de los eosinófilos tienen un núcleo denso en electrones y están rodeados por una matriz electrolúcida. Estos gránulos se componen de 4 proteínas: proteína básica mayor, integrada en la parte central; la peroxidasa de eosinófilos; la neurotoxina derivada de los eosinófilos y la proteína catiónica de eosinófilos. Dentro de los gránulos específicos de los eosinófilos también se encuentran varias quimiocinas preformadas, factores de crecimiento y citocinas, como IL-4, IL-2, GM-CSF, IL-5, IL-13, CCL5/regulado en la activación, linfocitos T normales expresados y secretados y eotaxina.
Los gránulos pequeños contienen fosfatasa ácida y arilsulfatasa. Por otra parte, las vesículas secretoras, también conocidas como microgránulos, tienen forma de mancuerna y contienen varios receptores, moléculas de adhesión y albúmina.
Los eosinófilos también contienen cuerpos lipídicos que se diferencian de los gránulos porque están rodeados por una monocapa de fosfolípidos. Los cuerpos lipídicos son los sitios de síntesis del leucotrieno y su formación es inducida en diversas condiciones inflamatorias experimentales así como en respuesta a diferentes estímulos.
Los eosinófilos usan una variedad de procesos de desgranulación para liberar selectiva o completamente su contenido celular. Estos procesos incluyen la exocitosis clásica (gránulos individuales que se fusionan con la membrana plasmática y descargan su carga); la exocitosis compuesta (varios gránulos se fusionan entre sí y con la membrana plasmática) y, poco a poco, se va produciendo la desgranulación (pequeños componentes de los gránulos se desprenden y se fusionan con la membrana plasmática) y finalmente, produciéndose la citólisis de los eosinófilos. La forma en que se regulan estos procesos aún se está investigando.
Por otra parte, los eosinófilos también liberan redes o trampas de ADN con contenido intacto de gránulos libres, siendo ésta una vía regulada de muerte clular en una trampa extracelular mediada por los eosinófilos, conocida como ETosis. Recientemente, la ETosis se asoció directamente con la formación de cristales de Charcot-Leyden.
| Función de los eosinófilos |
Se cree que los eosinófilos son células efectoras en la defensa del cuerpo contra las infecciones parasitarias, aunque su mecanismo de acción puede diferir dependiendo del parásito. Un mecanismo de acción propuesto es la liberación de proteínas granulares de los eosinófilos tóxicos a través de la ETosis. Otros mecanismos incluyen la muerte celular citotóxica dependiente de anticuerpos, que en la esquistosomiasis está realizada tanto por los eosinófilos como por los neutrófilos.
Los eosinófilos podrían usan mecanismos similares para causar daño tisular e inflamación en los eosinófilos asociados a enfermedades.
Sin embargo, los ratones con deficiencia de eosinófilos pueden eliminar algunas infecciones parasitarias, lo que sugiere que, al menos, hay redundancia en las defensas antiparasitarias del cuerpo.
Más recientemente, a los eosinófilos se les han atribuido funciones en el mantenimiento de las células plasmáticas de la médula ósea, en las respuestas de recuperación de las vacunas y en la modulación de una variedad de respuestas mediadas por las células T, así como funciones en la reparación de los tejidos, el metabolismo de la glucosa y las grasas y, tal vez, la vigilancia de los tumores. Muchos de estos estudios se han hecho en modelos animales y la extensión a los seres humanos aún no se ha llevado a cabo. Estudios murinos también sugieren que los eosinófilos pueden ser divididos en subconjuntos inflamatorios u homeostáticos y no está claro si estos existen en los seres humanos.
Los autores dan gran importancia al papel que representan los eosinófilos en la patogénesis de la enfermedad alérgica. Están físicamente presentes en las vías respiratorias de los pacientes con asma eosinofílica, dentro de los pólipos de aquellos con rinosinusitis crónica con pólipos, y en el tracto gastrointestinal (GI) de aquellos con enfermedad GI eosinofílica (EGIE). También pueden estar presentes en la piel de pacientes con erupciones cutáneas relacionadas con medicamentos.
| Definiciones de eosinofilia |
> Eosinofilia en sangre
En general, el grado de eosinofilia se define por el recuento absoluto de eosinófilos (número de eosinófilos circulantes en sangre periférica). El recuento absoluto de eosinófilos se puede determinar multiplicando el recuento total de glóbulos blancos por el porcentaje de eosinófilos. Su rango normal en sangre es de 0 a 500 células/mm3 y el porcentaje típico es <5% del recuento de leucocitos. Sin embargo, la presencia de eosinofilia no se puede determinar solo por el porcentaje porque la leucopenia conduce a un aumento relativo en el porcentaje de eosinófilos, y viceversa.
Los seres humanos muestran variación diurna en varios parámetros hematológicos, incluidos los recuentos de eosinófilos en sangre.
Un estudio reciente mostró una mediana variabilidad dentro del sujeto de 40 células/mm3, o 20%, con un pico ocurrido a la 1 AM, pero en aquellos con recuentos de eosinófilos dentro del rango normal, el máximo se produjo al mediodía. El cambio del recuento de eosinófilos dentro del mismo sujeto en 24 horas, en aquellos con eosinofilia se desconoce.
La eosinofilia se define como >500 eosinófilos/mm3. El grado de eosinofilia puede clasificarse como leve (500–1500 células/mm3), moderado (1500–5000 células/mm3), o grave (>5000 células/mm3). La hipereosinofilia se define como eosinofilia moderada a grave (≥1500 células/mm3). Para cumplir con la definición del SHE se requiere evidencia de daño de órgano blanco producido por la hipereosinofilia.
Comúnmente se cree que el grado de eosinofilia en la sangre se correlaciona con la gravedad de la enfermedad, pero no hay evidencia para apoyar esto. El daño de órganos terminales puede ocurrir con niveles moderados de eosinofilia. Un estudio reciente describe un grupo raro de personas con hipereosinofilia asintomática y sin evidencia de daño en el órgano diana a pesar de una evaluación exhaustiva y regular.
Algunos medicamentos (por ej., corticosteroides) y condiciones médicas transitorias (infección bacteriana) también pueden enmascarar un mayor grado de eosinofilia al suprimir temporalmente el recuento de eosinófilos absoluto. Por lo tanto, son importantes la evaluación clínica y el contexto.
> Eosinofilia Tisular
En condiciones homeostáticas, la mayoría de los eosinófilos residen en los tejidos, mayormente en el tracto GI excepto el esófago. También se encuentran en el timo, glándulas mamarias y útero.
La residencia tisular está regulada por la expresión de células reclutadoras de eosinófilos en los tejidos, como la eotaxina 1. En condiciones patológicas, los eosinófilos son reclutados fen otros sitios anatómicos, incluidos el pulmón, la piel y el esófago, y pueden aumentar más en sitios que normalmente ya contienen eosinófilos, como el estómago. Sin embargo, los umbrales para lo que se considera un aumento patológico no están bien definidos.
La experiencia con la eosinofilia esofágica muestra cómo eso podría cambiar. En 2007, para fines de investigación y atención clínica, se estableció un estándar de consenso multidisciplinario de médicos interesados en la esofagitis eosinofílica. En el contexto clínico y con los síntomas adecuados, a falta de respuesta a un ensayo de dosis elevada de inhibidores de la bomba de protones (IBP) (para tratar la enfermedad por reflujo gástrico), el tejido que muestra un recuento máximo de eosinófilos de 15 o más eosinófilos/campo de alta potencia, se acordó como el estándar mínimo para el diagnóstico de esofagitis eosinofílica. Este estándar estuvo a menudo acompañado por otras características histológicas, tales como la hiperplasia de la zona basal, el aspecto desgranulado de los eosinófilos y la presencia de microabscesos eosinófilos. Estudios posteriores revelaron una entidad descrita como sensible a los IBP.
La eosinofilia esofágica y estudios transcriptómicos sugieren que esta entidad se parece más a la esofagitis eosinofílica que a la enfermedad por reflujo gástrico. Por lo tanto, la mayoría de las guías actuales sugieren que no es necesario un ensayo con dosis elevadas de los IBP para establecer un diagnóstico de esofagitis eosinofílica si está presente el contexto clínico correcto. Podría considerarse a los IBP como un tratamiento inicial de la esofagitis eosinofílica, aunque todavía no se sabe si los pacientes con eosinofilia esofágica que responden a los IBP mantienen la capacidad de respuesta a este tratamiento. Más allá del esófago, los eosinófilos normalmente se encuentran en el tracto GI y, por lo tanto, pueden representar un papel en el mantenimiento de la homeostasis.
Hay pocos estudios que describen los niveles de eosinófilos en el tejido GI normal. Debrosse y col. realizaron un estudio en niños de un hospital pediátrico que fueron sometidos a endoscopia con biopsias y que finalmente resultaron ser normales. Como umbral para la eosinofilia tisular se propuso usar 2 veces el recuento máximo de eosinófilos en cada segmento GI. Este método da como resultado diferentes segmentos del intestino grueso con diferentes cortes, siendo el colon ascendente el que tiene el umbral más elevado para el diagnóstico.
En la actualidad, se acepta la existencia de eosinofilia gástrica si hay 30 eosinófilos/campo de gran aumento en al menos 5 campos de alta potencia, con fines de historia clínica e inscripción natural en el estudio, con un umbral similar para el intestino delgado. Se necesitan más estudios para determinar si solo el número de eosinófilos define la enfermedad en cada segmento del tracto GI , y hasta qué umbral.
| Interpretación de la eosinofilia |
El contexto clínico es importante. ¿El valor anormal es nuevo? ¿Es persistente? ¿Cuáles son los síntomas clínicos y los medicamentos concomitantes asociado con la anormalidad de laboratorio?, y ¿Cómo cambia eso a lo largo del tiempo y/o el tratamiento? Por lo tanto, una historia completa y un examen físico detallados son fundamentales en este entorno.
La eosinofilia leve en un paciente que toma dosis elevadas de corticosteroides mientras está febril se interpretaría de manera diferente a un paciente sin síntomas pero con una eosinofilia moderada. Por otra parte, si los eosinófilos son patógenos y causantes de enfermedades o si son parte del medio celular que se reclutan en el sitio de la enfermedad sigue siendo una pregunta abierta.
| Condiciones asociadas con eosinofilia |
La eosinofilia está asociada con varias condiciones médicas, incluyendo enfermedades alérgicas y condiciones específicas, desde las más comunes como la reacción a un medicamento, hasta enfermedades eosinofílicas raras, como los SHE.
> Infecciones
La eosinofilia se asocia clásicamente con enfermedades parasitarias (por ej., helmintiasis). Un enfoque es considerar las características del paciente, viajes previos e historial de exposición, con lo que se obtiene una guía para la evaluación. Si se sospecha que un paciente tiene una infección parasitaria, puede estar justificada su derivación a un infectólogo. La infección por el virus de la inmunodeficiencia humana (VIH) puede estar asociada con eosinofilia, aunque existen factores de confusión, como el uso de medicamentos o la concomitancia de infecciones oportunistas o parasitarias.
En un estudio de casos y controles, alrededor del 10% de los pacientes con VIH no tratados previamente tenían eosinofilia en sangre y una carga viral de ARN del VIH ligeramente más elevada, sin diferencias en edad, sexo, raza o recuento basal de CD4, en comparación con un grupo de pacientes control con VIH y sin eosinofilia en sangre ni tratamiento previo.
En ese estudio, la presencia de erupción, incluyendo la foliculitis eosinofílica (aunque no exclusivo de ella) fue más probable en pacientes con eosinofilia en sangre (46% en el grupo casos vs 25% en el grupo control). Las infecciones tuberculosas y micobacterianas no tuberculosas se asocian con eosinofilia. Cabe destacar que las infecciones bacterianas se asocian con eosinopenia.
El tratamiento está dirigido a la infección subyacente. La resolución de la infección suele asociarse con una disminución o resolución de la eosinofilia, aunque esto puede no ocurrir inmediatamente. Por ejemplo, después de una dosis única de dietilcarbamazina o del tratamiento con ivermectina se puede observar infección por loa loa, eosinofilia asociada al postratamiento, pudiendo tardar días o semanas en resolverse.
En un estudio de infección por Strongyloides stercoralis en zonas rurales de India, la eosinofilia disminuyó 6 meses después del tratamiento y, en algunos casos, no se resolvió. En raras ocasiones puede ocurrir el SHE como resultado de infecciones parasitarias activas mientras que el tratamiento en esos casos todavía está dirigido a la causa infecciosa subyacente.
> Medicamentos
Los medicamentos son la causa más común de eosinofilia persistente en los países desarrollados, pero el hallazgo de laboratorio no es ni sensible ni específico de una reacción a un fármaco. En un estudio de reacciones farmacológicas cutáneas agudas en pacientes hospitalizados (n = 55), la eosinofilia en sangre (definida como >700 células/mm3) se observó solo en el 18%, y se observaron eosinófilos tisulares en el 24% de los casos.
Por otra parte, solo la mitad de aquellos con eosinófilos tisulares comprobados por biopsia (12% de los casos) tenían eosinofilia concurrente en sangre. Por lo tanto, la falta de eosinofilia en la sangre o de eosinófilos detectados en la biopsia de tejido no debe ser usada para descartar alergia o reacción eosinofílica a fármacos.
Por otra parte, hay muchas reacciones alérgicas a medicamentos que no se cree que estén mediadas por eosinófilos o asociadas a ellos, incluidos la hipersensibilidad tipo 1 mediada por inmunoglobulina (Ig) E, la hipersensibilidad retardada (dermatitis de contacto), la enfermedad del suero y la necrólisis epidérmica tóxica/síndrome de Stevens- Johnson.
Las reacciones a los medicamentos asociadas a la eosinofilia pueden variar desde la eosinofilia benigna y transitoria con o sin erupción cutánea hasta una afectación más grave de los órganos internos como en la reacción a fármacos con eosinofilia y síntomas sistémicos (ESS).
En un estudio prospectivo de un solo centro para investigar las reacciones eosinofílicas a medicamentos en pacientes hospitalizados, la incidencia de reacciones farmacológicas asociadas a eosinófilos fue de 16,67/10.000 admisiones, siendo el 56% asintomáticos, el 13% con reacciones tisulares en la piel y tejidos blandos, el 7% con afectación visceral y 23% con una presentación clínica consistente con el ESS.
El cuadro ESS es una enfermedad potencialmente mortal.
Se presenta de forma tardía (semanas) después del inicio del fármaco, con síntomas que incluyen fiebre (90 a 100%), a menudo elevada, junto con una erupción morbiliforme. También puede haber hinchazón facial en la fase temprana. Estos síntomas pueden ir seguidos o acompañados por afectación visceral. Las 2 más comunes son la afectación hepática y la linfadenopatía, pero puede incluir miocarditis, colitis, neumonitis y trastornos del sistema nervioso central.
Los medicamentos comúnmente asociados con este síndrome son: alopurinol, sulfasalazina, antibióticos (ß lactámicos, minociclina, dapsona, sulfametoxazol, vancomicina), anticonvulsivos (lamotrigina, ácido valproico, carbamazepina, fenobarbital, fenitoína), agentes antirretrovirales (abacavir, nevirapina, raltegravir, efavirenz) y, ranelato de estroncio.
Se cree que la causa de la enfermedad es una combinación de células T CD81 activadas dirigidas contra el fármaco y los virus. Los hallazgos de laboratorio incluyen eosinofilia, cambios en el entorno de las citocinas T colaboradoras y reactivación viralꟷcomo ha sido detectado por la reacción en cadena de la polimerasa del virus del herpes humano (VHH) 6.
Recientemente se ha comenzado a analizar los factores asociados con fármacos o causas específicas. Los ejemplos incluyen la correlación del aumento del factor tímico sérico y la activación regulada por quimiocinas (TARC)/niveles de CCL17 en pacientes con ESS con reactivación del VHH6, y diferencias clínicas y de laboratorio entre aquellos con ESS provocado por lamotrigina vs. otros fármacos. Aunque hubo estudios que aislaron células T en sangre y piel para fármacos específicos de pacientes con ESS, actualmente no hay pruebas para determinar qué fármaco es el responsable.
El diagnóstico de ESS puede ser difícil debido a un cuadro clínico incompleto o a una presentación atípica, lo que llevó al desarrollo de sistemas de puntaje para el diagnóstico, con el fin de simplificar el enfoque. Un ejemplo es el puntaje RegiSCAR. El tratamiento de los casos leves de ESS incluye la suspensión del fármaco y medidas de apoyo. Las manifestaciones cutáneas se pueden tratar con corticoides tópicos. En los pacientes con afectación visceral, a menudo se utilizan dosis elevadas de corticosteroides. La recuperación completa después de la abstinencia del fármaco puede llevar semanas o meses.
Estudios retrospectivos previos reportan una mortalidad del 5-10%, mientras que un estudio prospectivo reciente informó 2 muertes entre 117 participantes, (1,7%) durante la fase aguda. Si bien cualquier medicamento puede provocar una reacción, se han reconocido que son más conocidos por causar reacciones específicas, como las descritas anteriormente para la ESS. En algunos casos, estas reacciones están ocasionadas por una susceptibilidad genética. Por ejemplo, las reacciones adversas cutáneas graves en respuesta al abacavir, alopurinol, carbamazepina y la nevirapina tienen más probabilidades de ocurrir en personas con alelos del antígeno leucocitario humano específico. (HLA).
Actualmente se recomienda la prueba de los alelos del HLA antes de iniciar la carbamazepina y abacavir. También es importante preguntarle al paciente sobre el uso de suplementos y medicamentos de venta libre.
El síndrome de mialgia eosinófila fue descrito después de la exposición a un contaminante en el suplemento L-triptófano a fines de la década de 1980, provocando engrosamiento de l piel, mialgias y otros compromisos orgánicos. Por otra parte, también se producen reacciones alérgicas a medicamentos que no están asociadas con eosinofilia ni mediadas por eosinófilos, como el síndrome de Stevens- Johnson.
> Malignidad
Una neoplasia oculta puede estar asociada con eosinofilia en sangre. En los pacientes con eosinofilia persistente de reciente aparición sin una causa clara y edad apropiada, se justifica evaluar la presencia de malignidad. La historia clínica y el examen físico completos pueden revelar síntomas y signos, como fiebre, escalofríos, pérdida de peso, linfadenopatía o esplenomegalia, para dirigir la evaluación específica.
La evaluación bioquímica podría revelar alteraciones en otros parámetros hematológicos (por ej., citopenias, células de aspecto displásico en el frotis). En estos casos se debe buscar una causa hematológica de la eosinofilia con derivación al especialista. Diversas formas de mastocitosis también se asocian con eosinofilia significativa pero, en esos casos, el tratamiento se adapta a la mastocitosis. Por otra parte, después de extirpar o tratar el tumor maligno, generalmente la eosinofilia se resuelve. En los trasplantes de células madre para el tratamiento de la malignidad, a veces puede observarse eosinofilia postrasplante como parte de injerto vs. enfermedad del huésped.
> Trastornos autoinmunitarios/desregulación inmunitaria
Varios trastornos autoinmunes se asocian con eosinofilia en sangre leve a moderada. En algunos casos, los eosinófilos también se hallan en el sitio de la enfermedad, como en la granulomatosis eosinofílica con poliangeítis (GEPA, antes conocida como Churg-Strauss). No está claro si los eosinófilos causan daño directo; si están intentando resolver la inflamación o son espectadores inocentes. En general, los tratamientos de las enfermedades autoinmunes con o sin eosinofilia en sangre son los mismos.
Un ensayo reciente de fase 3 de la terapia anti-IL-5 para tratar la GEPA sugiere que los propios eosinófilos juegan algún papel patogénico, porque hubo una disminución significativa en los brotes de la enfermedad en los pacientes tratados, a pesar de la reducción gradual de la terapia concomitante con corticosteroides, que apuntaría más allá de los eosinófilos. Actualmente, la terapia anti-IL-5 se considera aprobada para la GEPA. Todavía queda por establecer si la terapia dirigida a los eosinófilos sería útil en otras enfermedades autoinmunes.
> Enfermedades con eosinofilia sanguínea asociada
Las inmunodeficiencias primarias (IDP) se asocian con eosinofilia y, con menor frecuencia, esta asociación puede ser más evidente en la edad adulta, con una presentación clínica consistente en una variedad de trastornos autoinmunes. Una explicación para la correlación es que oligoclonal/repertorios de linfocitos restringidos, como los que se observan en varias IDP conducen a eosinofilia y esto fue demostrado experimentalmente en un modelo murino. Una vez más, el tratamiento definitivo es el dirigido a la IDP subyacente.
> Trastornos atópicos
La eosinofilia leve a moderada puede estar asociada con una amplia variedad de trastornos atópicos, incluyendo dermatitis atópica, rinitis alérgica y asma. La enfermedad gastrointestinal eosinofílica (EGIE) como la esofagitis eosinofílica se diagnostica por la eosinofilia tisular junto con el contexto clínico. En la esofagitis eosinofílica suele haber una eosinofilia sanguínea muy leve mientras que, en la gastroenteritis eosinofílica (compromiso del estómago o del intestino delgado), se ha hallado eosinofilia en sangre de moderada a grave, pero se están realizando estudios más amplios, necesarios para confirmar esos datos.
Muchas de estas enfermedades se tratan con corticoides (tópicos o sistémicos), que se dirigen tanto a los eosinófilos como a otras células inmunitarias, como los linfocitos, que podrían ser la población de células incitadoras. En el caso de la EGIE, las dietas empíricas de eliminación de alimentos también se han utilizado, particularmente en la población pediátrica, y en la esofagitis eosinofílica tienen una eficacia del 70-80%. Más recientemente, el tratamiento con terapias dirigidas a los eosinófilos en un subgrupo de pacientes con asma eosinofílica que usaron anti-IL-5 o anti-IL-5R dio como resultado un aumento de la función pulmonar y disminución de los brotes de enfermedades.
Ahora, ambos tipos de productos biológicos han sido aprobados para su uso como complemento de las terapias de mantenimiento en el asma grave con fenotipo eosinofílico (moderado a severo para anti–IL-5R). Este hallazgo sugiere que los eosinófilos representan un papel patogénico directo, al menos en ciertos endotipos de asma.
Por el contrario, el tratamiento con anti-IL-4RA (que bloquea la señalización de IL-4-IL-13) en aquellos con asma de moderada a grave dieron como resultado una mejoría clínica similar, pero se acompañó de aumentos transitorios de los recuentos de eosinófilos periféricos, así como de los niveles séricos de proteína de gránulos de eosinófilos. Con algunas excepciones individuales, la eosinofilia resultante no parecía causar daño o afectar la eficacia, aunque no se constató en un análisis de subgrupos que estratificó a los pacientes con diferentes grados de aumento de eosinófilos en sangre periférica.
En otras condiciones, la eosinofilia puede estar asociada con la embolización de colesterol, la irradiación y la insuficiencia suprarrenal. La eosinofilia puede estar asociada con la embolización de colesterol, la irradiación y la insuficiencia de adrenalina.
El SHE comprende a un grupo de enfermedades raras definidas por tener un recuento persistente de eosinófilos en sangre ≥1500 células/mm3 (durante al menos 4 semanas, salvo que fuera necesario un tratamiento inminente) y evidencia de daño de órganos diana resultante de esta eosinofilia. Esta definición ha evolucionado a lo largo de los años transcurridos desde que Chusid describió por primera vez la enfermedad. Una forma de subdividir este grupo puede ser en subtipos clínicos.
En el SHE mieloide hay una aberración genética en el linaje de las células eosinofílicas y/o mieloides, ésto acompañado por un aspecto displásico de los eosinófilos, aumento de los niveles séricos de vitamina B12 y triptasa y signos clínicos que podrían incluir esplenomegalia. En el SHE linfoide hay una población clonal aberrante de células T (a menudo CD3dimCD4 positivo) que secreta niveles elevados de IL-5, promoviendo así la eosinofilia.
A veces, los pacientes con eosinofilia restringida a un solo órgano como la EGIE tiene eosinofilia en sangre periférica y cumplen criterios de SHE, y estos pacientes se clasifican como SHE superpuesto o superposición del SHE. Este síndrome asociado es una categoría utilizada para el SHE que se desarrolla en presencia de una malignidad o infección parasitaria, o una reacción a un medicamento. El SHE familiar consta de escasas familias con herencia autosómica dominante de eosinofilia en sangre, y a menudo sin muchos síntomas.
Un pequeño grupo de pacientes muestra hipereosinofilia pero no tiene síntomas perceptibles o daño de órgano diana a pesar de su evaluación deliberada durante varios años. Se considera que estas personas tienen hipereosinofilia de significado desconocido y permanecen sin tratamiento pero monitoreados a lo largo de los años.
| Evaluación de la eosinofilia |
Además de la historia y el examen físico, sería útil obtener otro hemograma completo con fórmula leucocitaria (para buscar eosinofilia), un panel metabólico básico, prueba de función hepática y, un frotis de sangre periférica para examinar si hay displasia celular. Según el contexto clínico (y la urgencia) y las causas probables, el estudio para la eosinofilia difiere y, a veces, se justifica la derivación a un subespecialista.
Sería razonable derivar a un hematólogo/oncólogo a los pacientes mayores con linfadenopatía, fiebre, pérdida de peso y citopenias.
En los pacientes con eosinofilia se debe evaluar la asociación con una larga historia de síntomas GI, como disfagia, especialmente un gastroenterólogo y un alergólogo/inmunólogo con experiencia en EGIE. Por otra parte, los hombres jóvenes con evidencia reciente de disfunción cardíaca y eosinofilia grave deben ser evaluados con urgencia para descartar el SHE mieloide, mediante biopsia de médula ósea y pruebas para los cambios genéticos causantes, incluyendo la translocación FIP1L1-PDGFRA.
Las pruebas de detección incluyen la determinación de triptasa sérica, vitamina B12, IgE sérica y citometría de flujo para células T aberrantes, así como pruebas de diagnóstico por imágenes, cardíacas y pulmonares, si está indicado. No hay pruebas validadas para determinar los desencadenantes alimentarios de la EGIE en la enfermedad GI, además de repetir un muestreo invasivo de tejido y, por lo tanto, no es útil medir un panel de alimentos IgE o IgG4 específicos.
Medir la IL-5 sérica no ha sido útil hasta ahora para determinar la causa de la eosinofilia, aunque es un predictor de respuesta a la terapia anti-IL-5 en un subconjunto de pacientes con síndrome hipereosinofílico. Por otra parte, hay pocos datos que indican que el nivel sérico de IL-5 se ve afectado por medicamentos como los corticosteroides. Las 4 proteínas granulares de los eosinófilos (MBP, EDN, EPO, ECP) se pueden medir en la sangre y otros fluidos y tejidos corporales, pero las correlaciones exactas, tanto con el diagnóstico como con la actividad de la enfermedad, aún no se han determinado. Por otra parte, la eosinofilia en sangre aislada y asintomática puede ser el signo más temprano de SHE.
Los estudios iniciales para los SHE deben incluir la evaluación del SHE mieloide si el recuento absoluto de eosinófilos aumenta notablemente (>5.000 células/mm3) con análisis de sangre (triptasa, vitamina B12), pruebas genéticas para mutaciones asociadas como FIP1L1/PDGFRA, y evaluación de la médula ósea. Si el contexto clínico sigue siendo preocupante (paciente masculino con niveles elevados de triptasa/B12, presencia de esplenomegalia y eosinófilos displásicos) a pesar de las pruebas genéticas negativas, vale la pena derivar a un especialista en SHE porque se han hallado negativos falsos.
Debido a que los síntomas pueden ocurrir sin previo aviso, es útil monitorear periódicamente el aumento del recuento de eosinófilos en ausencia de síntomas o evidencia de daño de órgano terminal, con historia y examen físico trimestral o anual, acompañado de pruebas dirigidas. No hay recomendaciones de consenso publicadas en cuanto al intervalo y modalidades de la prueba porque los pacientes pueden tener tipos muy diferentes de órganos diana afectados.
Es razonable realizar análisis de sangre anuales, pruebas de función pulmonar y ecocardiografía en aquellos con hipereosinofilia, especialmente en los primeros 5 años. En aquellos con SHE superpuesto (por ej., EGIE, granulomatosis eosinofílica con poliangeítis), los aumentos en los recuentos de eosinófilos pueden preceder o acompañar a un brote de enfermedad y esos recuentos a menudo regresan a la línea de base del paciente con tratamiento.
Si los síntomas que acompañan a ese brote de enfermedad son diferentes o nuevos, se debe hacer una evaluación específica de esos hallazgos para dilucidar si están causados por un nuevo proceso patológico o relacionado con los eosinófilos.
| Guías futuras: papel de la terapia dirigida a los eosinófilos |
En los últimos años se ha demostrado que la terapia dirigida a los eosinófilos reduce los brotes de la enfermedad y ha logrado la aprobación de la FDA (Administración de Drogas y Alimentos) de EE. UU. para el asma eosinofílica grave (anti-IL-5, mepolizumab [100 mg por vía subcutánea] y reslizumab [peso establecido]; anti–IL-5R, benralizumab) y la granulomaatosis eosinofílica con poliangfeítis (mepolizumab 300 mg por vía subcutánea).
En casos de SHE refractario con riesgo de vida, el mepolizumab en altas dosis (300 a 700 mg) puede ser apropiado aplicándolo al estudio de uso compasivo (NCT00244686). Un pequeño estudio de fase 2 de benralizumab (anti–IL-5R) para pacientes con SHE grave mostró la supresión de la eosinofilia periférica y la mejoría sintomática durante 48 semanas en el 74% de los pacientes. Todavía se necesitan ensayos multicéntricos de fase 3 más grandes para ambos productos biológicos en el tratamiento del SHE.
Los fármacos dirigidos a otros receptores de membrana de eosinófilos o que reducen el recuento de eosinófilos sin un mecanismo definido están siendo evaluados en diversas condiciones asociadas a los eosinófilos.
Si los productos biológicos IL-5/IL-5R u otras terapias dirigidas a los eosinófilos serán útiles en otras enfermedades agudas o crónicas asociadas a eosinófilos sigue siendo un enigma. Por ejemplo, ¿Serían útiles en los cuadros agudos de ESS? ¿Podrían bloquear la eosinofilia y las reacciones postratamiento durante el tratamiento de ciertas helmintiasis? Estas son preguntas inexploradas.
Hay más de una década de datos de seguridad en el uso de la terapia anti-IL-5 para el SHE de un estudio de uso compasivo. Sin embargo, se desconocen los efectos de la depleción de los eosinófilos a largo plazo con la terapia anti-IL-5R (benralizumab) y puede ser diferente porque esta terapia parece resultar en un agotamiento más completo de los eosinófilos, incluidos los residentes en tejidos.
Por otra parte, es posible que los eosinófilos no sean patogénicos en todas las condiciones con las que están asociados. Los autores esperan que en la a próxima década, el uso de estos nuevos productos biológicos dirigidos a los eosinófilos revele en cuál enfermedad la reducción del nivelo de eosinófilos es beneficiosa.
Resumen y comentario objetivo: Dra. Marta Papponetti