Las anemias microcíticas se caracterizan por la producción de glóbulos rojos más pequeños de lo normal. El tamaño pequeño de estas células se debe a la disminución de la producción de hemoglobina, el principal componente de los eritrocitos.
Las causas de la anemia microcítica son:
- La falta de globina (talasemia).
- La menor liberación de hierro para el grupo hem de la hemoglobina (anemia de la inflamación)
- La falta de suministro de hierro al grupo hem (anemia por deficiencia de hierro)
- Los defectos de la síntesis del grupo hem (anemias sideroblásticas).
Talasemias
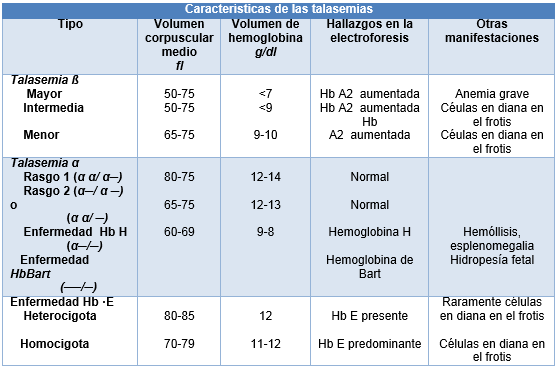
Las talasemias son enfermedades de la síntesis de hemoglobina, con subtipos que se denominan según la cadena de hemoglobina afectada. Dado que cada cromosoma 16 lleva 2 copias del gen que codifica la cadena α, hay 4 tipos de α-talasemia rasgo 1, rasgo 2, enfermedad por hemoglobina H y, enfermedad por hemoglobina Bart. Los pacientes con rasgo talasémico no tienen anemia o la misma es muy leve, con grados variables de microcitosis ─más pronunciada en los pacientes con las características del rasgo 2.
La supresión de la o las mutaciones en 3 genes de la cadena α provoca la enfermedad por hemoglobina H, en la cual la anemia es más notoria, a menudo con un componente hemolítico. La enfermedad por hemoglobina de Bart se caracteriza por la falta de producción de la cadena α, dando como resultado la hidropesía fetal y debida a la falta de producción de hemoglobina fetal y adulta.
Las principales localizaciones geográficas de la talasemia α son África, la zona del Mediterráneo y el sudeste asiático, pero las formas más graves─ la enfermedad por hemoglobina H y la enfermedad por hemoglobina de Bart─se hallan solo en la zona del Mediterráneo y el sudeste asiático.
La razón de esta asociación geográfica con las forma más graves es la presencia de las 2 formas moleculares del rasgo 2 de la talasemia α ─en una forma, cada cromosoma tiene una copia del gen mutada (trans) y en la otra, un cromosoma tiene mutados ambos genes (cis). El genotipo predominante es la forma trans, pero la forma cis se encuentra en otras áreas y puede dar lugar a la enfermedad por hemoglobina H y la enfermedad por hemoglobina Bart.
La talasemia β es común en el área mediterránea y el sudeste de Asia. Debido a que en el cromosoma 11 hay una copia de la cadena β de la hemoglobina, los pacientes pueden ser heterocigotas (talasemia menor) u homocigotos (talasemia mayor) para la cadena de hemoglobina defectuosa. Algunos pacientes son homocigotas para las mutaciones en la, pero conservan la síntesis de la cadena β residual, lo que resulta en un fenotipo intermedio (talasemia intermedia).
Los pacientes con talasemia menor presentan anemia microcítica leve. La talasemia mayor es una enfermedad grave y se manifiesta poco después del nacimiento con necesidad de transfusiones. Como su nombre lo indica, la talasemia intermedia tiene una presentación variada, desde la anemia dependiente de transfusiones hasta la anemia ligeramente más grave la de los pacientes con talasemia menor.
En el sudeste asiático también es común la enfermedad por hemoglobina E, en la que la lisina está sustituida por la glutamina en la posición 26 de la cadena β. Esta mutación también activa el sitio de empalme de un ARN mensajero (ARNm) alternativo que conduce a una marcada reducción en la síntesis de proteínas. Las personas heterocigotas para la hemoglobina E tienen microcitosis con células en diana, y los homocigotas presentan anemia leve. Sin embargo, los niños con una copia de cada uno de los genes de la talasemia β y del gen de la hemoglobina E tienen un fenotipo grave de anemia, dada su dependencia de las transfusiones.
Anemia de la inflamación
Los estados inflamatorios suelen acompañarse de anemia microcítica, la cual tiene un origen dual. Uno, es que la producción renal de eritropoyetina está suprimida por las citocinas inflamatorias, dando lugar a la producción de eritrocitos más pequeños. El otro es la falta de hierro disponible para el desarrollo de los eritrocitos lo que provoca la microcitosis.
La falta de hierro se debe en gran parte a la proteína hepcidina, un reactante de fase aguda que reduce la absorción del hierro y su liberación de los depósitos corporales. La proteína ferroportina es la mediadora del eflujo celular de hierro. La hepcidina se une a la ferroportina y la regula hacia abajo, de modo que bloquea el hierro absorbido por los enterocitos e impide que entre en la circulación. Al mismo tiempo, impide la liberación de hierro de sus depósitos corporales para ser utilizado en el desarrollo de los glóbulos rojos.
Deficiencia de Hierro
Comúnmente, la deficiencia de hierro provoca anemia, que es la más común de las anemias. Además de representar un papel importante como portador de oxígeno en el grupo hem de la hemoglobina, el hierro se halla en muchas proteínas celulares esenciales, como los citocromos y la mioglobina, por lo que no es raro que la falta de hierro tenga otros efectos además de la anemia.
Tres estudios se han ocupado de investigar la fatiga provocada por la deficiencia de hierro, en ausencia de anemia. Dos de ellos mostraron que en mujeres con un nivel de ferritina <50 ng/ml, el suplemento de hierro por vía oral redujo la fatiga sin modificar significativamente el nivel de hemoglobina. El tercer estudio halló una disminución de la fatiga con la administración de hierro parenteral en mujeres con un nivel de ferritina ≤15 ng/ml, o una saturación de hierro ≤20%.
Debido a la pérdida de hierro obligada a través de la menstruación, las mujeres están en mayor riesgo de deficiencia de hierro que los hombres. La pérdida de hierro promedio en todas las mujeres es de 1 a 3 mg por día, mientras que la cantidad de hierro proveniente de los alimentos suele ser inadecuada para mantener un balance positivo de hierro.
Un estudio de 1967 mostró que el 25% de los participantes, mujeres universitarias sanas, no tenía reservas de hierro en la médula ósea y el 33% tenía poco hierro en los depósitos. El embarazo aumenta la demanda de hierro, requiriendo más de 6 mg/día gasta el final del embarazo.
Los atletas son otro grupo de riesgo de deficiencia de hierro. El origen de la pérdida de hierro es la sangre en el tracto gastrointestinal y la hemólisis por ejercicio que conduce a la pérdida urinaria de hierro. En la deficiencia de hierro también interviene la menor absorción digestiva, porque los niveles de hepcidina suelen estar elevados en los atletas que sufren inflamación inducida por el entrenamiento físico. Aunque es obvio que la anemia manifiesta puede alterar el desarrollo de la actividad física, cada vez hay más evidencia de que la deficiencia de hierro sin anemia también puede ser determinante.
La obesidad y sus tratamientos quirúrgicos también conllevan el riesgo de ferropenia. Los pacientes obesos suelen ser deficientes en hierro, con niveles aumentados de hepcidina, lo que también disminuye la absorción. Después de la cirugía bariátrica, la incidencia de deficiencia de hierro alcanza el 50%. Debido a que el sitio principal de la absorción del hierro es el duodeno, las cirugías de derivación que sortean el duodeno se asocian con mayor incidencia de deficiencia de hierro. Sin embargo, esta deficiencia se considera una secuela de la mayoría de las derivaciones de la cirugía bariátrica.
Diagnóstico
La presencia de microcitosis puede ser la pista para hallar la etiología de la anemia, ya que un valor <70 fl es raro en los pacientes con anemia de la inflamación. Se ha propuesto una variedad de métodos predictivos como el uso de los índices hemáticos para diferenciar la talasemia de la deficiencia de hierro, pero su poder predictivo es limitado y por lo tanto se requieren otras pruebas específicas. En el frotis de sangre los microcitos se pueden reconocer por ser más pequeños que el núcleo de los linfocitos.
También se puede observar hipocromía—el aumento del área de palidez central de los glóbulos rojos. En la anemia ferropénica y la anemia de la inflamación predominan las células microcíticas, pero en la talasemia β y la enfermedad por hemoglobina E, también hay células en diana.
Los pacientes con talasemia β menor tienen un nivel de hemoglobina de10-13 g/dl y el volumen corpuscular medio es de 65 a 75 fl. Estos pacientes tienen mayor producción de hemoglobina con cadenas δ (hemoglobina A2), la que habitualmente está aumentada en la electroforesis. El problema es que la existencia concurrente previa de deficiencia de hierro puede atenuar el aumento de la hemoglobina A2 en la mayoría de los pacientes.
La talasemia α no tiene expresión electroforética. El diagnóstico puede hacerse por exclusión en un paciente con microcitosis y anemia leve o sin anemia ni deficiencia de hierro. El diagnóstico preciso requiere el análisis del ADN. La presencia de hemoglobina H (un tetrámero de las cadenas β) en la electroforesis junto con microcitosis severa es diagnóstica de enfermedad por hemoglobina H, en la que también puede haber hemólisis y esplenomegalia.
En la actualidad, el diagnóstico de anemia de la inflamación es por exclusión. El diagnóstico se apoya en 3 hallazgos: un valor de eritropoyetina inapropiadamente elevado en respuesta a la anemia, con preservación de la función renal y sin alteración de los depósitos de hierro, sin otra causa de anemia. En estos pacientes, la saturación de hierro es baja y la capacidad de unión total del hierro es baja a normal. En el futuro, será muy valioso disponer ampliamente del análisis de la hepcidina, ya que su elevación apoya el diagnóstico de anemia de la inflamación.
A lo largo de los años se han propuesto muchas pruebas para el diagnóstico de la deficiencia de hierro, entre ellas la medición de la ferritina, la que en la actualidad es la prueba de mayor eficacia y rentabilidad dadas las deficiencias de las otras pruebas.
En la deficiencia de hierro grave, el volumen corpuscular medio es bajo, pero la coexistencia de enfermedades como las hepatopatías puede hacer que los de los eritrocitos no sean tan pequeños. En la anemia por deficiencia de hierro y la talasemia, el contenido de hemoglobina de los reticulocitos es bajo aunque también está reducido en la anemia de la inflamación, reflejando la dificultad en la liberación del hierro para ser utilizado en el desarrollo de los eritrocitos.
En la anemia de la inflamación, el hierro sérico está bajo y falsamente elevado con la ingesta de hierro. El aumento de la capacidad de unión del hierro es específico de la deficiencia de hierro, pero debido a que esa capacidad está disminuida en la inflamación, el envejecimiento y la mala nutrición, su sensibilidad es baja. La saturación de hierro es baja tanto en la deficiencia de hierro como en la anemia de la inflamación.
En los pacientes ferropénicos, los niveles séricos del receptor de transferrina soluble están elevados en, no así en los pacientes con anemia de la inflamación. Sin embargo, los niveles pueden elevarse en los pacientes con cualquier enfermedad asociada a un aumento de la masa eritrocítica, como las anemias hemmolíticas o la eucemia linfocítica crónica. La manera más segura de hacer el diagnóstico de anemia por deficiencia de hierro es la tinción del hierro en la médula ósea, pero es un método invasivo y caro.
Aunque la transcripción del ARNm de la ferritina es regulada hacia arriba por la inflamación, la síntesis de ferritina está regulada por el contenido de hierro celular, y el ARNm de la ferritina es trasladado a la proteína solamente cuando la célula se llena de hierro.
Por lo tanto, con la inflamación, el paciente que posee una cantidad adecuada de hierro puede tener un nivel de ferritina muy elevado mientras que es raro que en los pacientes con deficiencia de hierro el nivel de ferritina sea >100 ng/ml. El límite inferior normal depende del estado clínico.
Un nivel de ferritina de 15 ng/ml es muy específico de la deficiencia de hierro, pero en los ancianos o en los pacientes con un estado inflamatorio, la deficiencia de hierro no puede ser descartada hasta que el nivel de ferritina no sobrepase los 100 ng/ml. Guyatt et al. comprobaron que la relación de posibilidad para la deficiencia de hierro es positiva con un nivel de ferritina de 40 ng/ml en ausencia de inflamación, y hasta 70 ng/ml si hay inflamación. Aunque imperfecto, la medición de la ferritina sérica es la prueba que más posibilidad tiene de brindar información acerca del estado férrico del paciente pero para interpretar el resultado hay que tener en cuenta la edad y el estado clínico.
La otra prioridad esencial ante la deficiencia de hierro es determinar la causa. Dado que no hay mecanismos naturales (salvo la menstruación) que provoquen la pérdida el hierro corporal siempre hay que asumir que la falta de hierro se debe a su pérdida. Cuando el origen de la pérdida no es evidente, se deben buscar lesiones en el tracto gastrointestinal, debido al elevado porcentaje de pacientes que tienen un sitio identificable donde se origina el sangrado.
Tratamiento
Talasemia
En los niños con formas graves de talasema, las transfusiones crónicas permiten un crecimiento y desarrollo normales. Sin embargo, si no se hace la quelación del hierro se desarrolla una insuficiencia endocrina, y la mayoría esos pacientes morirá en la segunda o tercera década de la vida debido a la sobrecarga de hierro. El tratamiento quelante permite prevenir o retardar las complicaciones.
El mejor tratamiento es el trasplante de células madre: los pacientes jóvenes sufrirán menos complicaciones que con otros tratamientos, y si el trasplante es exitoso no es necesario continuar con la terapia de transfusiones y la quelación.
El tratamiento de los pacientes con talasemia intermedia o enfermedad por hemoglobina H es más difícil debido a la variedad de presentaciones. En los pacientes que dependen de las transfusiones es esencial la quelación del hierro, ya que tienen mayor absorción del hierro con el peligro de sobrecarga, la que también puede producirse en los pacientes cuya dependencia de las transfusiones es mínima.
Los pacientes con talasemia no requieren un tratamiento específico. Sin embargo, en el caso de la maternidad es necesario determinar el volumen corpuscular medio en la pareja, pues si es <75 fl está indicado hacer un estudio genético más específico.
Anemia de la inflamación
La terapia más efectiva para la anemia de la inflamación es eliminar la causa subyacente, pero en muchos pacientes esto no se logra. Debido a que los niveles de eritropoyetina están bajos se han usado agentes estimulantes de la eritropoyesis con buenos resultados, ya que aumentan el recuento de eritrocitos, pero su utilización está limitada por su elevado costo y problemas de seguridad. En los modelos animales, el bloqueo de la hepcidina disminuye la anemia.
Deficiencia de hierro
Tratamiento con hierro oral
El medicamento más utilizado para la deficiencia de hierro es el sulfato ferroso, en dosis de 325 mg (65 mg de hierro elemental) por vía oral, 3 veces/día. En varios ensayos se ha comprobado que las dosis bajas de hierro elemental (15-o 20 mg/día) son tan efectivas como las dosis más elevadas, y con menos efectos colaterales. La razón puede ser la saturación de la absorción de hierro en los enterocitos; una dosis de hierro puede bloquear la absorción de las dosis siguientes.
El consumo de carne también ayuda a la absorción del hierro de las proteínas de la carne. El calcio y las fibras pueden disminuir la absorción, lo que puede contrarrestarse con la ingestión de vitamina C. Un potente inhibidor de la absorción del hiero es el té, el cual puede reducirla en un 90%. También lo hace el café pero en menor medida. En cuanto al hierro de la dieta, la tasa de absorción del hierro proveniente de fuentes hem es 10 veces más elevada que la de origen no hem.
Un tratamiento conveniente para aportar hierro por vía oral es comenzar con 325 mg diarios de sulfato ferroso (la forma más barata) con las comidas que contienen carne. Evitar el té y el café y tomar vitamina C (500 unidades con el comprimido de hierro, 1 vez/día), para ayudar a la absorción. Si hay intolerancia al sulfato ferroso se puede administrar 325 mg de gluconato ferroso (35 mg de hierro elemental).
La respuesta satisfactoria se evidencia por el aumento del número de reticulocitos al cabo de la primera semana, y del nivel de hemoglobina en la segunda semana del tratamiento, el cual debe continuar hasta llenar los depósitos. En algunos pacientes hay varios factores que explican la falta de respuesta al tratamiento oral. Primero, los efectos gastrointestinales (epigastralgia, constipación) pueden atentar contra el cumplimiento del paciente.
Estos síntomas adversos pueden mejorar disminuyendo la dosis. Segundo, en un paciente que ha sangrado (por ej., por enfermedad intestinal inflamatoria), la pérdida de hierro puede ser demasiado grande para poder compensarla por vía oral. Finalmente, la absorción del hierro puede disminuir en presencia de enfermedad celíaca o cirugía intestinal.
Administración parenteral de hierro
Para los pacientes que no responden adecuadamente al tratamiento hay varias opciones intravenosas. El hierro parenteral mejora los depósitos de hierro sin problemas de absorción o efectos adversos gastrointestinales. Su mayor desventaja es la reacción a la infusión.
El hierro con dextran de alto peso molecular se asocia a una tasa de reacción significativamente mayor que otras formas de hierro parenteral y no se recomienda su utilización. La sucrosa de hierro y el gluconato férrico son productos de reciente aparición con tasas de reacción más bajas, pero para llenar completamente los depósitos se requieren infusiones frecuentes. Cada vez hay más evidencia de que el hierro en dextran de bajo peso molecular tiene una tasa de reacciones similar a la de los productos más nuevos, pero permite administrar dosis más elevadas de hierro—hasta 1.000 mg—en una sola sesión.
El ferumoxitol es un óxido de hierro superparamagnético recubierto con carbohidrato que se comercializa como agente para el reemplazo de hierro y también como un agente de contraste para la resonancia magnética. La única complicación es la hipotensión grave, observada en el 1,9% de los pacientes en estudios post comercialización. Por otra parte, si dentro de los 3 meses de haber recibido el fármaco se necesitan estudios de resonancia magnética, los radiólogos deben tener en cuenta que el paciente ha recibió erumoxitol. Otra opción es la carboximaltosa férrica.
El hierro parernteral está indicado en cualquier caso de deficiencia de hierro refractaria al hierro oral. El hierro en dextran de bajo peso molecular puede ser la opción más cara y más conveniente. Para los pacientes con reacciones de hipersensibilidad al hierro dextran hay otras opciones. Los pacientes con deficiencia de hierro por sangrado─por ej., por telangiectasia hemorrágica hereditaria—pueden necesitar hierro en infusión.

El futuro
Al ser una enfermedad genética, la talasemia sigue siendo el objetivo ideal de la terapia génica. Hay varios ensayos clínicos en marcha, y varios pacientes ya han sido sometidos al tratamiento, con algunos signos preliminares de buenos resultados.
También hay cada vez mayor interés por elevar los niveles de hemoglobina fetal, con el fin de mejorar la anemia en los pacientes con talasemia β mayor, y sobre todo para las personas con talasemia intermedia. Un tratamiento muy prometedor para la anemia de la inflamación es la manipulación de la vía de la hepcidina.
Aunque se han hecho grandes progresos, aún queda mucho por dilucidar sobre el metabolismo del hierro, incluyendo el receptor de la absorción del hierro hem. Finalmente, falta todavía investigar el papel de los nuevos marcadores─como los polimorfismos en una proteína muy importante sensora del hierro, la proteasa serina transmenmbrana 6, que puede aumentar el riesgo de deficiencia de hierro.
Traducción y resumen objetivo: : Dra. Marta Papponetti