Autor/a: Dres. Landolfi R, Marchioli R, Kutti J, Gisslinger H N Engl J Med. 2004 Jan 8;350(2):114-24
En general, la trombocitosis se descubre en forma incidental como una anormalidad de laboratorio, al realizar el hemograma por una indicación no relacionada. Sin embargo, al comprobarse la trombocitosis aparece un desafío diagnóstico importante. La trombocitosis suele ser un proceso reactivo (trombocitosis secundaria) o estar ocasionada por un trastorno clonal de la médula ósea (mieloproliferativa); esta última categoría incluye la trombicitemia esencial. Es muy difícil diferenciar ambos tipos de trombocitosis guiándose por los hallazgos clínicos o de laboratorio. Existen diferencias fundamentales entre ellas, en cuanto a su causa, fisiopatología y manifestaciones clínicas.
Mecanismos de la trombocitosis
La trombopoyetina es la hormona principal en la regulación de la diferenciación y la proliferación de los megacariocitos, aunque en el proceso también pueden intervenir varias citocinas como las interleucinas 6 y 11. Los megacariocitos y su progenie plaquetaria tienen receptores para la trombopoyetina, conocidos como c-Mpl. En el plasma, la trombopoyetina se una al c-Mpl en la superficie de las plaquetas circulantes; la trombopoyetina restante no ligada queda para promover la proliferación de los megacariocitos.
Por lo tanto, cuando el recuento de plaquetas disminuye, los niveles plasmáticos de trombopoyetina libre ascienden para estimular la megacariocitopoyesis; en cambio, cuando el recuento de plaquetas aumenta, los niveles reducidos de trombopoyetina hacen más lenta la megacariocitopoyesis. De este modo, la cantidad total de plaquetas (y de megacariocitos) puede regular la producción de plaquetas y mantenerlas en un estado estable. En algunos casos de trombocitosis reactiva, un estímulo inflamatorio subyacente puede regular hacia arriba la producción de trombopoyetina en el hígado.
En la trombocitosis reactiva (secundaria), los niveles plasmáticos de trombopoyetina son elevados o inapropiadamente normales. En casos de inflamación aguda, esta elevación precede al incremento del recuento plaquetario. Los niveles plasmáticos de interleucina 6 también están elevados en la trombocitosis reactiva: esta interleucina, la cual representa un papel importante en la fase aguda de la respuesta inflamatoria y las enfermedades neoplásicas, regula hacia arriba la expresión del RNA mensajero (RNAm) de la trombopoyetina en el hígado. Por lo tanto, la interleucina 6 puede ser un mediador clave de la síntesis aumentada de trombopoyetina y la consecuente trombocitosis reactiva.
Los niveles de trombopoyetina también están elevados o inapropiadamente normales en la trombocitosis clonal, pero en este caso, el mecanismo comprende las anormalidades en la regulación de la captación de la trombopoyetina sintetizada constitutivamente mediada por el receptor c-Mpl. En la trombocitemia esencial, un defecto clonal en la expresión del c-Mpl de las plaquetas y los megacariocitos provoca la alteración en la unión de la trombopoyetina y, en consecuencia, hay niveles mayores que los esperados de trombopoyetina plasmática libre.
Esta situación contrasta con la de otros trastornos mieloproliferativos, en los cuales la proliferación clonal de un linaje hematopoyético provoca, por un mecanismo de retroalimentación fisiológico, la supresión de los factores de crecimiento específicos que normalmente controlan la diferenciación y la proliferación de los linajes hematopoyéticos individuales: por ejemplo, los niveles de eritropoyetina sérica están disminuidos en la policitemia vera, mientras que en la leucemia mieloide crónica, están disminuidos los niveles del factor estimulante de las colonias de granulocitos.
En los trastornos mieloproliferativos, la unión de la trombopoyetina a los megacariocitos está disminuida debido a la disminución de la cantidad y la función de los receptores de la trombopoyetina, pero en la trombocitemia esencial, esos progenitores también están muy hipersensibles a la acción de la hormona. Esto provoca la mayor proliferación de los megacariocitos y de la producción de plaquetas observadas.
Trombocitosis reactiva (secundaria)
En general, la causa más común de trombocitosis en la población médica general es un proceso reactivo o secundario. El grado de elevación del recuento plaquetario no permite diferenciar claramente la trombocitosis reactiva de la clonal. En una serie de 732 pacientes clínicos y quirúrgicos con recuento de plaquetas de 500.000/mm3 o más, 643 (88%) tenían una trombocitosis reactiva; las causas subyacentes más frecuentes fueron el daño tisular por cirugía mayor, infección, cáncer e inflamación crónica. Del mismo modo, en una serie de 280 pacientes hospitalizados consecutivos con recuentos plaquetarios de 1 millón/mm3 o más, 231 (82) tenían trombocitosis reactiva, 11 (4%) trombocitosis de causa desconocida y solo 38 (14%) presentaban un trastorno mieloproliferativo.
La trombocitosis reactiva deriva de la elevación de los niveles endógenos de trombopoyetina, interleucina 6, otras citocinas o, las catecolaminas que pueden estar producidas por procesos inflamatorios, infecciosos o neoplásicos o, también, en situaciones de estrés.
En la mayoría de los pacientes aparecen síntomas clínicamente significativos de una enfermedad sistémica activa subyacente. Sin embargo, en otros, la trombocitosis secundaria puede estar causada por trastornos subclínicos como el cáncer oculto. Este último grupo de pacientes es el que presenta el problema diagnóstico más difícil para el clínico. Antes de hacer el diagnóstico de trombocitosis clonal (mieloproliferativa), cuyo diagnóstico casi siempre es por exclusión y tiene consecuencias terapéuticas diferentes, el clínico debe confirmar que el recuento plaquetario elevado no se debe a una enfermedad subyacente oculta pero potencialmente tratable.
Trombocitosis familiar
Los casos raros de trombocitosis familiar fueron inicialmente descritos como un trastorno autosómico dominante en el cual las mutaciones con actividad funcional del gen de la trombopoyetina provocan la hiperproducción de trombopoyetina y una elevación notable de sus niveles plasmáticos. Estas formas de trombocitosis familiar son un ejemplo del mecanismo genético de la enfermedad descrito recientemente, el que se caracteriza por la pérdida de la inhibición traslacional, provocando una mayor eficacia de la traslación del mRNA. Sin embargo, en la actualidad también se reconocen otros modos de herencia, en los cuales los niveles de trombopoyetina son normales, de manera que, desde el punto de vista genétic,o la trombocitosis familiar es un trastorno heterogéneo. Además, dado que estos pacientes pueden tener complicaciones trombóticas y vasculares, es posible que algunos casos de trombocitemia esencial esporádica sean formas familiares.
Trombocitosis clonal
La trombocitemia esencial es uno de los trastornos mieloproliferativos crónicos, un grupo de trastornos relacionados con la policitemia vera, la leucemia mielógena crónica y, la metaplasia mieloide con o sin mielofibrosis. La trombocitosis no se produce exclusivamente en la trombocitemia esencial, sino que también puede ocurrir en otros trastornos mieloproliferativos, en particular la policitemia vera. Por otra parte, algunos casos de trombocitemia esencial en realidad representan una entidad de reciente identificación que es la mielofibrosis prefibrótica, la cual evoluciona hacia la mielofibrosis manifiesta. La trombocitosis también puede asociarse con el sindrome mielodisplásico 5q, caracterizado por la deleción del brazo largo del cromosoma 5 y es uno de los sindromes denominados mielodisplásico y mieloproliferativo mixtos. El recuento de plaquetas elevado en la trombocitemia esencial y otros trastornos mieloproliferativos está provocado por la trombocitosis clonal
Clonalidad
La clonalidad de estos trastornos ha sido demostrada utilizando marcadores polimórficos ligados a X en pacientes mujeres. Estudios recientes demostraron que la trombocitemia esencial no siempre es clonal. Estos hallazgos destacan la posibilidad de que la enfermedad no clonal pueda evolucionar hacia la enfermedad clonal, que la clonalidad está restringida en algunos pacientes al linaje megacariocítico o, que la trombocitemia esencial es un trastorno heterogéneo. Debido a la dificultad en la interpretación de los ensayos de clonalidad, estas pruebas no tienen aplicación clínica con fines diagnósticos.
La trombocitemia esencial sigue siendo un diagnóstico de exclusión de las causas secundarias (reactivas) de trombocitosis. Las complicaciones hemorrágicas y trombóticas son las principales causas de enfermedad y muerte en los pacientes con trombocitemia esencial y otros trastornos mieloproliferativos, en particular en los pacientes ancianos con factores de riesgo asociados. Estos problemas hemostáticos no ocurren en la trombocitosis secundaria, independientemente del grado en que esté elevado el recuento de las plaquetas, a menos que un trastorno sistémico subyacente predisponga al paciente.
Se cree que su ausencia en la trombocitosis secundaria se deba al hecho de que la interacción de las plaquetas con la pared vascular sigue siendo cualitativamente normal. En la trombocitosis clonal, las complicaciones hemorrágicas tienden a ser del "tipo plaquetario," en forma de hemorragia espontánea en sitios superficiales (la piel o las membranas mucosas de los tractos gastrointestinal, respiratorio o genitourinario). Los pacientes con trombocitosis esencial que tienen trombocitosis extrema (1.5 millones/mm3) tienen mayor riesgo de sangrado, por su consumo de aspirina u otros fármacos antiplaquetarios.
En una serie de 187 pacientes consecutivos con trombocitosis esencial seguidos en una sola institución, el 50% tuvo al menos un episodio trombótico dentro de los 9 años que siguieron al diagnóstico. La isquemia microvascular de los dedos es característica de la trombocitosis esencial. Puede asociarse con el sindrome de eritromelalgia, caracterizado por dolor urente o ardor distribuidos en áreas de las manos y los pies, en particular la superficie plantar. Los signos físicos pueden estar ausentes o incluir calor, y eritema de coloración oscura y moteado en las áreas involucradas. Debido a que la oclusión por plaquetas se caracteriza por comprometer solo a la microvasculatura, los pulsos periféricos pueden permanecer palpables. En la trombocitosis esencial, la isquemia digital no tratada puede provocar la gangrena.
En la trombocitosis clonal, las complicaciones neurológicas pueden estar ocasionadas por la isquemia cerebrovascular mediada por las plaquetas. Esto ocurre en el 25% de los pacientes con trombocitosis esencial y pueden manifestarse por síntomas inespecíficos, como la cefalea o los mareos crónicos, o con signos neurológicos focales. La trombosis arterial o venosa de los vasos grandes también puede complicar el curso de la trombocitosis clonal. La trombosis de las venas profundas y el embolismo pulmonar son las formas más comunes de trombosis venosa por trombocitosis clonal. Sin embargo, las complicaciones trombóticas intraabdominales, como la trombosis de la vena hepática (sindrome de Budd-Chiari) y la trombosis de la vena porta, son características de los trastornos mieloproliferativos. El aborto espontáneo recurrente y el retardo del crecimiento fetal afectan a cerca del 50% de los embarazos en las mujeres con trombocitemia esencial. Estas complicaciones se caracterizan por infartos placentarios múltiples causados por la trombosis plaquetaria.
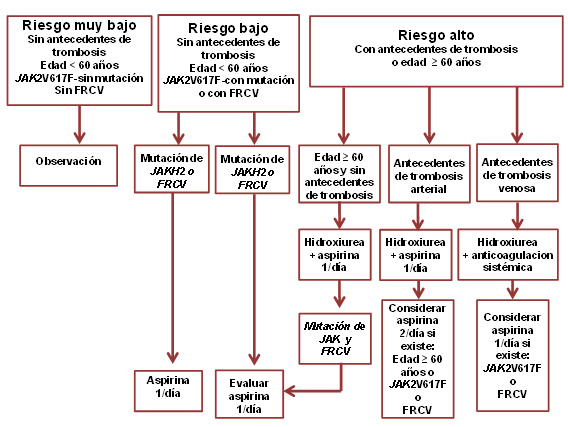
Aunque el grado de elevación del recuento plaquetario no se correlaciona con el riesgo de trombosis, su control mediante la citorreducción disminuye la frecuencia de trombosis en algunos pacientes. Las anormalidades cualitativas de las plaquetas y los leucocitos o la disfunción endotelial también pueden contribuir con las complicaciones hemostáticas de los trastornos mieloproliferativos. Otros factores de riesgo de complicaciones trombóticas en pacientes con trombocitemia esencial son la edad avanzada, el antecedente de trombosis, la hipercolesterolemia y, el tabaquismo.
Historia natural
Aunque el análisis precoz de la supervivencia actuarial entre los pacientes con trombocitemia esencial no puso de manifiesto una disminución significativa de su expectativa de vida, un estudio poblacional más reciente, el Olmsted County Study, demostró que la supervivencia entre los pacientes con trombocitemia esencial fue significativmanete peor que entre los controles sanos comparables por edad y sexo. Las complicaciones trombóticas y vasculares son las causas principales de muerte en los pacientes con trombocitemia esencial; sin embargo, en algunos pacientes (que no han recibido recientemente tratamiento leucemogénico) la enfermedad termina por convertirse en una leucemia aguda o una mielodisplasia o, puede evolucionar hacia la mielofibrosis.
Diagnóstico diferencial
No existen hasta el momento hallazgos diagnósticos que permitan distinguir la trombocitosis secundaria de la clonal. Como expresaron antes lo autores, los pacientes con trombocitosis secundaria sufren una enfermedad sistémica responsable del aumento de las plaquetas. A diferencia de los pacientes con trombocitosis secundaria, los pacientes con trombocitosis clonal tienen complicaciones trombóticas, vasculares y hemorrágicas. El 40% de los pacientes con trombocitemia esencial tienen esplenomegalia pero también puede ocurrir en algunos pacientes con trombocitosis secundaria.
Las pruebas de laboratorio tampoco permiten la diferenciación. En la trombocitosis clonal, y no así en la trombocitosis secundaria, en el frotis de sangre periférica suelen encontrarse plaquetas gigantes. En la trombocitosis clonal también existen anormalidades variadas de la función plaquetaria, pero no en la trombocitosis secundaria. Estas anormalidades pueden incluir el sindrome de von Willebrand adquirido y la ausencia de agregación plaquetaria inducida por la epinefrina. El frotis y la biopsia de la médula ósea muestran el aumento de los megacariocitos en ambas formas de trombocitosis, pero existen algunas diferencias leves en su morfología. En la trombocitosis secundaria, los megacariocitos tienen aspecto normal, mientras que en la trombocitosis clonal pueden ser gigantes y displásicos, con poliploidia, y pueden asociarse con grandes masas de detritos plaquetarios. Por lo tanto, el examen de la médula ósea es un examen útil en la trombocitosis clonal, aunque no sea diagnóstico.
Ninguna de las formas de la trombocitosis tiene anormalidades citogenéticas diagnósticas. Sin embargo, algunos pacientes con trombocitemia esencial tienen el cromosoma Filadelfia o el reordenamiento BCR-ABL, aun en ausencia de leucocitosis u otras manifestaciones de la leucemia mieloide crónica. Aunque las consecuencias clínicas de estos hallazgos no están del todo esclarecidas, algunos de estos pacientes con trombocitemia esencial (con trombocitosis aislada) en realidad, tienen una variante de la leucemia mieloide crónica. Aunque estas pruebas no son aplicables en la clínica, el desarrollo continuado de ensayos de clonalidad y pruebas de expresión del c-Mpl en los megacariocitos y las plaquetas prometen ser herramientas diagnósticas que permitan diferenciar la trombocitosis secundaria de la clonal.
En los pacientes con trombocitosis secundaria no es necesario el tratamiento para disminuir las plaquetas o la administración de agentes antiplaquetarios, porque su anormalidad plaquetaria no los pone en riesgo de eventos hemostáticos o vasculares. Sin embargo, es muy importante identificar la causa de su trombocitosis secundaria, aun cuando no tenga manifestaciones clínicas, para que el tratamiento esté dirigido a la enfermedad subyacente. Una eritrosedimentación y un nivel de proteína C reactiva normales pueden descartar un proceso inflamatorio. La investigación de un cáncer oculto debe incluir el examen físico, la búsqueda de sangre oculta en la materia fecal, una radiografía de tórax y otras pruebas indicadas por los síntomas y los signos, localizados y sistémicos.
Por el contrario, en la trombocitosis clonal puede ser necesario disminuir el número de plaquetas. En la actualidad, todavía no se cuenta con estudios que evalúen la eficacia profiláctica de esta medida en los pacientes con trombocitemia esencial, asintomáticos y con riesgo bajo, independientemente del grado de trombocitosis. Si los pacientes tienen antecedentes de trombosis o sangrado, factores de riesgo cardiovascular o más de 60 años, el riesgo de cuadros trombóticos y vasculares es elevado. Un estudio prospectivo, aleatorizado y controlado demostró que el tratamiento citorreductor de las plaquetas con hidroxiurea ofrece beneficios a los pacientes de alto riesgo con trombocitemia esencial y un aumento en la tasa de supervivencia libre de trombosis. Los pacientes con trombocitemia esencial con isquemia cerebrovascular o digital activa deben recibir cuanto antes tratamiento con agentes citorreductores de las plaquetas. La plaquetaféresis se reserva para los pacientes complicados con un accidente cerebrovascular agudo o una isquemia digital aguda, consiguiendo una rápida mejoría.
Excepto en algunos ancianos que no toleran otros fármacos, los agentes alquilantes no se usan más para reducir las plaquetas porque pueden causar leucemia aguda. Hasta hace poco, un agente alquilante, la hidroxiurea, era el reductor del número de plaquetas de elección por su uso sencillo. Sin embargo, se le atribuyen efectos leucemogénicos, sobre todo con su uso prolongado, combinada con otros fármacos o, en pacientes con trombocitemia esencial con deleción del brazo corto del cromosoma 17.
En este momento, el tratamiento de la trombocitosis clonal se hace con anagrelida, un derivado de la quinazolina de administración oral que inhibe la proliferación y la diferenciación de los megacariocitos, siendo la opción de primera línea para el tratamiento reductor del número de plaquetas. No tiene potencial leucémico y por lo tanto es útil en los pacientes jóvenes con trombocitemia esencial que requieren un control plaquetario a largo plazo. La dosis inicial es 2 mg/día (divididos en 2 a 3 dosis) y puede aumentarse a razón de 0,5 mg/día, cada 7 días, hasta conseguir el recuento de plaquetas esperado; dosis máxima, 10 mg/día. Casi el 30% de los pacientes no tolera la anagrelida por sus propiedades vasodilatadora e inotrópica positiva; sus efectos colaterales son la retención de líquido, las palpitaciones y las arritmias, la insuficiencia cardíaca y, las cefaleas, debiendo tener un cuidado especial en los ancianos o cardiópatas. Dichos efectos disminuyen con el tiempo pero muchos pacientes desarrollan anemia.
El interferón alfa es un agente efectivo no mutagénicos que reduce el número de plaquetas pero está limitado por sus efectos colaterales graves que lo hacen intolerable para el 20% de los pacientes. En las mujeres en edad fértil, es de elección, dado que la hidroxiurea es teratogénico y la angrelida atraviesa la placenta. En ciertas mujeres jóvenes con trombocitosis clonal puede aplicarse el trasplante de células madre hematopoyéticas en estado avanzado o con complicaciones.
La aspirina puede ser muy efectiva en la trombocitemia esencial con complicaciones trombóticas recurrentes, en particular cuando existe isquemia digital o cerebrovascular, combinada con el tratamiento reductor de plaquetas. Sin embargo, debe ser usada con precaución en otros pacientes con trombocitosis clonal porque puede causar hemorragia. Se ha comprobado que 100 mg de aspirina diarios son efectivos para prevenir las complicaciones trombóticas de la policitemia vera sin aumentar el riesgo de hemorragia. Con respecto a un agente antiplaquetario nuevo, el clopidogrel, los datos de su aplicación en la trombocitosis clonal son escasos.